Dr Rajiv Desai
An Educational Blog
What Causes Cancer
What Causes Cancer:
_
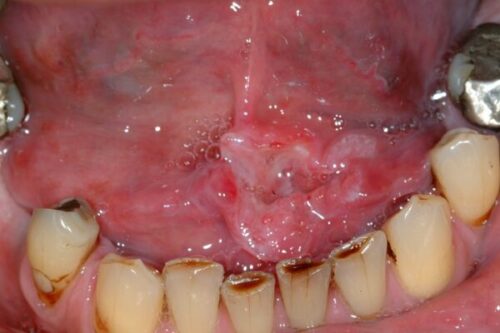
Figure above shows squamous cell carcinoma of the left anterior floor of mouth.
_
Section-1
Prologue:
International Cancer Institute has defined Cancer as a term used for diseases in which abnormal cells divide without control and can invade nearby tissues. Cancer is the second leading cause of death worldwide. Globally annually, there were an estimated 20 million new cases of cancer and 10 million deaths from cancer or nearly one in six deaths. The cancer burden will increase by approximately 60% over the next two decades, further straining health systems, people and communities. It’s often said that cancer is not one, but many diseases. In fact, there are more than 200 forms of cancer, and all of these are caused by cells in the body acquiring the ability to grow and proliferate uncontrollably. Deaths from cancer are usually caused by secondary tumours, which form when cancer cells spread to various parts of the body in a process known as metastasis. More than a third of people experience a form of cancer; and breast, lung, prostate, and bowel cancers are some of the most common kinds.
_
Cancer is a genetic disease caused by accumulation of DNA mutations and epigenetic alterations leading to unrestrained cell proliferation and neoplasm formation. Many these alterations involve subtle sequence changes in DNA (i.e., mutations) and other involve epigenetic alterations (epimutations). The somatic mutations may originate as a consequence of random replication errors or exposure to carcinogens (e.g., radiation) and can be exacerbated by faulty DNA repair processes. Cancer cells acquire their ability to multiply unchecked through DNA mutations, and they continue to mutate and evolve as the disease develops. This means that, even within a certain type of cancer, one person’s tumour can be genetically different to another person’s, and this can make it difficult to choose the best treatment for each individual patient. While most cancers arise sporadically, clustering of cancers occurs in families that carry a germline mutation. You may be born with a genetic mutation that you inherited from your parents. This type of mutation accounts for a small percentage of cancers. Most gene mutations occur after you’re born and aren’t inherited. A number of forces can cause gene mutations & epimutations, such as smoking, radiation, viruses, cancer-causing chemicals, obesity, hormones, chronic inflammation and a lack of exercise.
_
The earliest case of cancer that researchers have so far been able to document occurred in a hominin (early human ancestor) whose remains date back 1.7 million years. Investigators located these remains in a South African cave, and they yielded evidence of osteosarcoma, an aggressive type of bone cancer, at the dawn of the human race. Yet humans and their ancestors are not the only animals to have been affected by cancer through history. Anecdotally, cancer is the leading cause of death in cats and dogs, and some birds, reptiles, and fish — in captivity and in the wild — can experience cancer, too. Furthermore, according to recent discoveries, even dinosaurs sometimes developed cancer.
_
In medicine, the earliest written description of diseases and cancer, a breast cancer, is found in the Edwin Smith Papyrus that was written approximately 3000 BC. The writer concluded that bulging tumor of the breast was a grave disease and there was no treatment for it. The Ebers Papyrus, dated circa 1500 BC, contains the first reference to a soft-tissue tumor, a fatty tumor, and includes reference to possible cancers of the skin, uterus, stomach, and rectum. The Egyptians attempted to treat tumors and cancers with cautery, knives, and salts, and introduced arsenic paste that remained in use as “Egyptian ointment” until the 19th century. The Sumerians, Chinese, Indians, Persians, and Hebrews of the same epoch were partial to herbal remedies such as tea, fruit juices, figs, and boiled cabbage, but in advanced cases, they did not hesitate to resort to solutions and pastes of iron, copper, sulphur, and mercury. Many of these concoctions remained in external and internal use, in various concentrations, for more than 3000 years.
_
Cancer is caused by genetic changes leading to uncontrolled cell growth and tumor formation. The basic cause of sporadic cancers is DNA damage and genomic instability due to environmental factors. A minority of cancers are due to inherited genetic mutations. Cancer is generally not contagious in humans, though it can be caused by oncoviruses and few bacteria. The term “environmental”, as used by cancer researchers, refers to everything outside the body that interacts with humans. The environment is not limited to the biophysical environment (e.g., exposure to factors such as air pollution or sunlight), but also includes lifestyle and behavioral factors. Over one third of cancer deaths worldwide (and about 75–80% in the United States) are potentially avoidable by reducing exposure to known factors. Common environmental factors that contribute to cancer death include exposure to different chemical and physical agents (tobacco use accounts for 25–30% of cancer deaths), environmental pollutants, diet and obesity (30–35%), infections (15–20%), and radiation (both ionizing and non-ionizing, up to 10%). These factors act, at least partly, by altering the function of genes within cells. Typically many such genetic changes are required before cancer develops. Aging has been repeatedly and consistently regarded as an important aspect to consider when evaluating the risk factors for the development of particular cancers. Many molecular and cellular changes involved in the development of cancer accumulate during the aging process and eventually manifest as cancer. Understanding the relative contribution of modifiable risk factors to cancer burden and their trends over time is crucial to informing cancer control efforts both locally and globally. The most common preventable risk factors for cancer are tobacco smoking or chewing, diet (low in fruits and vegetable and high in fatty foods, red meats, etc.), obesity, and alcohol. While doctors have an idea of what may increase your risk of cancer, cancers do occur in people who don’t have any known risk factors. It could be random replication error purely by chance. However, cells contain a mechanism that recognizes when a mistake occurs and repairs the mistake. Occasionally, a mistake is missed. This could cause a cell to become cancerous.
_
Research on causes of cancer is critical to progress against the disease. Cancer can be caused by many things, including exposure to cancer-causing substances, certain behaviors, age, and inherited genetic mutations. Studying the causes of cancer helps researchers understand the process by which normal cells are transformed into cancer cells and identify genetic, environmental, and behavioral risk factors for cancer. This knowledge can lead to new ways of preventing and treating the disease. Research on the causes of cancer also creates opportunities to improve public health, not only by identifying cancer risk factors in populations, but also by providing data that regulatory agencies can use to set safety standards or reduce exposure to toxins that are found to be associated with cancer. Researchers use many different approaches to identify potential causes of cancer, from cell-based and animal studies to human observational studies. Research in basic cancer biology can reveal the mechanisms by which biological, chemical, and physical carcinogens initiate and promote cancer. Genetic analyses, such as genome-wide association studies, exome sequencing, and whole-genome sequencing, allow researchers to identify genetic changes that may be associated with cancer risk. Epidemiological approaches—including cohort studies, case-control studies, exposure-assessment studies, family studies, and genomic studies—are used to identify possible causes of cancer and study the patterns of risk in large populations. My endeavour is to study what causes cancer so that we can find cure.
_____
_____
Abbreviations and synonyms:
APC = Antigen presenting cell
CD = Cluster of differentiation
CSF = Colony-stimulating factor
CTLA = Cytotoxic T lymphocyte-associated molecule
CXCL = Chemokine (C-X-C motif) ligand
DC = Dendritic cell
IL = Interleukin
CSC = Cancer stem cell
MSC = Mesenchymal stem cell
ESC = Embryonic stem cell
SSC = Somatic stem cell
EMT = Epithelial-to-mesenchymal transition
BCL-2 = B-cell lymphoma 2
p53 = Tumour-suppressor protein p53
TERT = Telomerase reverse transcriptase
VEGF = Vascular endothelial growth factor
EGF = Epidermal growth factor
FGF = Fibroblast growth factor
TAM = Tumor-associated macrophages
CRC = Colorectal cancer
RCC = Renal cell carcinoma
mTOR = Mammalian Target of Rapamycin
AMPK = Adenosine monophosphate (AMP)-dependent kinase
HCA = Heterocyclic amines
PAH = Polycyclic aromatic hydrocarbons
CIS = Cervical carcinoma in situ
CIN = Chromosomal instability
MMR = Mismatch repair
HER2 = Human epidermal growth factor receptor 2
MANAs = Mutation-associated neoantigens
TME = Tumor microenvironment
TSA = Tumor-specific antigen
TAA = Tumor-associated antigen
ADCC = Antibody-dependent cell-mediated cytotoxicity
CDC = Complement-dependent cytotoxicity
MAP/ERK = Mitogen-activated protein/Extracellular signal-regulated kinase
MAPK = Mitogen-activated protein kinase
MHC = Major histocompatibility complex
NK = Natural killer
PI3K-Akt = Phosphatidylinositol-4,5-bisphosphate 3-kinase- AKT8 virus oncogene cellular homolog
STAT = Signal transducer and activator of transcription
TCR = T cell receptor
TGF-β = Transforming growth factor beta
Th1 = Type 1 T helper
Treg = Regulatory T cell
PD-1 = Programmed cell death receptor-1
PD-L1 = Programmed cell death ligand-1
RSTK = Receptor serine-threonine kinase
RTK = Receptor tyrosine kinases
______
______
Section-2
Introduction to cancer:
Neoplasia is the uncontrolled, abnormal growth of cells or tissues in the body, and the abnormal growth itself is called a neoplasm or tumor. It can be benign or malignant. Benign neoplasms tend to grow slowly; displace, but do not tend to invade, the surrounding body tissues; and do not spread throughout the body. Malignant neoplasms, on the other hand, can be unpredictable and grow at various rates (sometimes rapidly), invade the tissues around them, and spread, or metastasize, to other parts of the body. The word “tumor” or “mass” is often used to describe the actual swelling or other physical appearance of a neoplasm. The word “cancer” is often confused with neoplasia, but only malignant neoplasms are truly cancers. Cancer is a genetic disease caused by accumulation of DNA mutations and epigenetic alterations leading to unrestrained cell proliferation and neoplasm formation. Cancer develops due to DNA mutations and epigenetic alterations that accumulate randomly. Cancer refers to any one of a large number of diseases characterized by the development of abnormal cells that divide uncontrollably and have the ability to infiltrate and destroy normal body tissue. Cancer often has the ability to spread throughout your body.
_
According to the definition provided by International Cancer Institute, a tumor is a non-specific term which refers to an abnormal mass of tissue that results when cells divide more than they should or do not die when they should. Tumors can be benign (innocent) or malignant (cancer), and it is important to carry out a biopsy of the affected tissue in order to decide if a suspicious growth is benign or malignant. Benign tumors are innocent or non-cancerous tumors which are usually localized and do not spread to different parts of the body and result in metastasis. Most benign tumors respond well to treatment, but if left untreated can give rise to serious complications due to their increased size, e.g., pressure and obstructive signs and symptoms. Benign tumors can also mimic malignant tumors, so it is important to make a correct diagnosis as soon as possible since earliest interventions are definitely helpful in avoiding fatal scenarios. Malignant tumors, on the other hand, are cancerous and could be fatal. They are usually resistant to treatment, grow rapidly and spread to various parts of the body, especially to lungs and liver and often recur even after the complete removal due to the aggressive cellular division. Not all tumors are fatal. Some oncologists suggest the removal of benign tumors due to possible complications like recurrence and obstructive or pressure symptoms, disrupting the patient’s lifestyle. However, the type of surgery and the time of intervention will be decided based on the location and size of the tumor.
_
The human body is made up of trillions of cells, about 100 trillion to be precise. Cells are the tiny building blocks of our tissues and organs. We all started life as a single cell. That cell made an internal copy of itself (replication) and then divided into two cells. Those two cells then also replicated and divided, so the two cells became four cells. The four cells replicated as well and divided into eight cells, and so on. Eventually these cells form a complete human being. Early cells, also called Stem cells, are undifferentiated. This means that they have the potential to become any kind of cell, but haven’t specialised. Once a certain number of cells have been formed, new cells begin to differentiate, and become unique cells, with very a specific purpose. Cells come in around 200 different types, and each has a special role in the body. They have different sizes and shapes, and even different life spans. When cells die or are damaged, they need to be replaced, so cell division is a process that continues throughout our lives. Each day, the human body makes about 300 billion new cells. More than half of these are red blood cells, which only last for around 120 days, and are constantly being replaced. This is a completely normal process. With a couple of important exceptions, when healthy cells divide, they are pre-programmed to become a specific sort of cell, and there are controls about whether they can replicate themselves. They also have a programmed age limit, after which they naturally die and are replaced.
Cells specialize to perform particular tasks. Some cells will cluster together to form a finger, for example. Others create skin and heal the skin when it is wounded. Cells get old and die after a certain amount of time (“programmed cell death,” or apoptosis), and replication ensures that new cells are made to take their place. When they are acting normally, cells “know” which other cells to join up with and stick to – and they also know when to stop replicating and die. Each type of cell has a particular role and set of knowledge or instructions in their DNA (genes). Our cells know how to make the right number of fingers on our hand for us (and they know that fingers should only grow on our hands). Each finger is covered with skin and each finger has a fingernail. If we cut our finger, the skin cells will start replicating and create new skin to heal the wound. If we lose a fingernail, our cells can grow a new one. But the cells will not create extra fingers, even if we lose one. The rules are clear for those cells, and they keep to the rules.
Because of billions of new cells made every day, there are inevitably going to be mistakes from time to time. A tumour occurs when there is a mistake in the copying process, and the replicated cell is faulty. The worst mistake is that they continue to replicate themselves without any limit. In addition, they are often unspecialised or undifferentiated, so they don’t serve a purpose, and they do not go through normal cell life cycles. Because the cells keep dividing, they can eventually form a lump, or mass, which affects the part of the body where they live. This body part may then be unable to work properly, or may not be able to work at all.
_
And any cell in the body has the potential to become cancerous if:
- it can divide;
- it builds up mistakes in certain genes; and
- these faults cause it to grow out of control.
Even though there are many boxes to tick, accumulating DNA damage is the most fundamental part. Some of this damage is avoidable – such as that caused by smoking and drinking alcohol – while some isn’t, such as the damage that comes from getting older. So even if we try and live as healthily as we can, many cells in our bodies still have the potential to become cancerous because of ageing, where our cells get worn down and don’t work as well over time.
Now we’ve covered what gives cells the potential to become cancerous, what stops all cells turning bad?
Our cells come with inbuilt safety systems that can stop cancer developing.
Some of these systems stop our precious DNA from becoming damaged in the first place. For example, the sun’s UV rays can cause a build-up of oxygen-containing molecules inside cells that can damage DNA. This is why our cells make antioxidants to mop them up.
But even with this safety system in place, genetic mistakes can still crop up and, in fact, do so relatively regularly. After all, each time a cell divides it copies the 3 billion DNA ‘letters’ that make up its genetic code per haploid set of chromosomes – a mammoth task to do error-free. And if a cell is exposed to lots of a cancer-causing substance, such as tobacco smoke, then its defences can be overwhelmed and DNA damage becomes inevitable.
That’s why cells also have proofreading machines that scan our DNA code, looking for faults and calling on molecular repair teams to fix any damage that’s found.
These very effective systems have evolved over billions of years, but they’re not fool proof, and mistakes can slip through the net. Sometimes a subtle genetic change might go unnoticed, or there could be so much damage that the repair machinery can’t cope. But even if this happens, there are further checks in place to stop the damaged cell from dividing and potentially leading to a cancer. For example, the cell can be forced to commit suicide so that it can’t pass on its faulty DNA to new cells.
But even then, on rare occasions, some damaged cells can slip past these checks.
That’s when the immune system swoops in, spying on cells that appear out of the ordinary and wiping them out. Frustratingly, cancer cells have evolved their own ways to defend from immune attack, such as dressing up in molecules that form an invisibility cloak. Ultimately, these can allow cancerous cells to evade destruction and go on to develop into the disease. But researchers are turning these tricks around and using them to their advantage, in the form of the latest immunotherapy treatments that can target certain cancers.
All in all, this paints a complex picture of how cancer starts. There are many things that can give a cell the potential to become cancerous, but there are checks in place to stop that from happening.
The more we learn about these processes, the more opportunities arise to intervene and attack cancer in ever more sophisticated and precise ways. That’s why understanding the basic biology of cancer is so fundamental to making progress against it.
_
What is Cancer?
International Cancer Institute has defined Cancer as a term used for diseases in which abnormal cells divide without control and can invade nearby tissues. Cancer cells can also spread to other parts of the body through the blood and lymph systems. If cancer cells start replicating, they don’t behave like normal cells. For example, they don’t know when to stop replicating and when to die. And they don’t always stick together, so they might break away and move through the blood vessels or lymphatic system and start growing somewhere else in the body. This process is called metastasis. This tumour that is metastatic and the primary tumour are both the same cancer type. For example, if breast cancer travels to the lungs, it is metastatic breast cancer and not lung cancer. Metastatic cancer is considered advanced cancer. Simply put, metastatic cancer is when cancer has moved or spread from its place of origin to a different part of the body.
All cells within the body have a purpose. Red blood cells bind to oxygen and deliver it to other cells in the body. Epithelial cells come together to produce skin that protects us from outside elements. Neurons enable thought and the sharing of information within the brain, followed by nerve impulses throughout the body. Regardless of their purpose, different cells have different typical lifespans. However, some cells gain an unusual capability—immortality. Cells that have obtained immortality do not die, but they lose their basic functions in the process. Unfortunately, these cells can come from anywhere in the body and form the basis of cancer.
_
What’s wrong with Cancer Cells?
Cancer cells behave differently than normal cells in the body. Many of these differences are related to cell division behavior. Cancer cells demonstrate a variety of unusual characteristics when grown in culture; two such examples are a lack of contact inhibition and a reduced dependence on the presence of growth factors in the environment. In contrast to normal cells, cancer cells do not cooperate with other cells in their environment. They often proliferate indefinitely in tissue culture. Cancer cells can multiply in culture (outside of the body in a dish) without any growth factors, or growth-stimulating protein signals, being added. This is different from normal cells, which need growth factors to grow in culture. Cancer cells may make their own growth factors, have growth factor pathways that are stuck in the “on” position, or, in the context of the body, even trick neighboring cells into producing growth factors to sustain.
Cancer cells also ignore signals that should cause them to stop dividing. For instance, when normal cells grown in a dish are crowded by neighbors on all sides, they will no longer divide. Cancer cells, in contrast, keep dividing and pile on top of each other in lumpy layers. The environment in a dish is different from the environment in the human body, but scientists think that the loss of contact inhibition in plate-grown cancer cells reflects the loss of a mechanism that normally maintains tissue balance in the body.
Another hallmark of cancer cells is their “replicative immortality,” a fancy term for the fact that they can divide many more times than a normal cell of the body. In general, human cells can go through only about 40-60 rounds of division before they lose the capacity to divide, “grow old,” and eventually die. Cancer cells can divide many more times than this, largely because they express an enzyme called telomerase, which reverses the wearing down of chromosome ends that normally happens during each cell division.
Cancer cells are also different from normal cells in other ways that aren’t directly cell cycle-related. These differences help them grow, divide, and form tumors. For instance, cancer cells gain the ability to migrate to other parts of the body, a process called metastasis, and to promote growth of new blood vessels, a process called angiogenesis (which gives tumor cells a source of oxygen and nutrients). Cancer cells also fail to undergo programmed cell death, or apoptosis, under conditions when normal cells would (e.g., due to DNA damage). In addition, emerging research shows that cancer cells may undergo metabolic changes that support increased cell growth and division.
_
The main differences between malignant (cancerous) and benign (non-cancerous) tumors are that malignant ones can
-spread into the surrounding tissue,
-destroy the surrounding tissue, and
-cause other tumors to develop.
Malignant tumors can be life-threatening. But there are also some kinds of cancer that develop so slowly in older people that they don’t lead to any problems in their lifetime. Benign tumors usually don’t cause much damage and aren’t normally life-threatening. But there’s no guarantee: Benign growths can become dangerous if they grow a lot, or they might become malignant after a certain amount of time.
These kinds of tumors start to create their own blood vessels so that they can continue to grow. The blood vessels supply them with extra oxygen, glucose (sugar) and hormones. This process of developing a blood supply system is called angiogenesis (the growth of new blood vessels). Once a tumor does this, it can start to invade the surrounding tissue. This is called invasive cancer. Active cancer cells can enter the bloodstream or lymphatic system and travel to other parts of the body. There they start the process of forming a tumor all over again somewhere else (metastatic or secondary cancer/tumor).
When a malignant tumor is contained within one area and hasn’t spread to the surrounding tissue, the medical term is “carcinoma in situ.” If this tumor stops growing, doctors say it is dormant (“dormant cancer cells”).
_
Your body repairs and replaces millions of cells every day. With such a high turnover rate, mistakes are unavoidable. But those cells are usually removed from your system because there’s something wrong about them that our body recognizes, just like your body would recognize if it were infected with a virus. White blood cells attack the altered cell and keep it from replicating. Occasionally, an altered cell is able to disguise itself from the immune system. This happens through a sequence of events where the cancer cells meet up with immune cells, target the immune cells’ receptors that recognize cancerous cells and systematically turn them off. Without the immune response, those cells replicate out of control, forming tumors.
_
Cancer is when abnormal cells divide in an uncontrolled way. Some cancers may eventually spread into other tissues. At its heart, cancer is the result of uncontrolled cell growth. Our bodies are composed of trillions of cells, all working together. In cancer, one of those cells stops paying attention to the normal signals that tell cells to grow, stop growing or even to die. Cancer cells still share many of the same needs and properties of normal cells but they become independent of the controls that make our body function smoothly. The process by which a normal cell changes into one that behaves so abnormally can take a long time and is often triggered by outside influences. There are several changes that must occur for a normal cell to become a cancer cell. Additional changes are needed for that single abnormal cell to form a group of cancer cells, called a tumor, and then for that tumor to grow and spread.
_
Cancer is actually a general term that describes a large group of related diseases. Every case of cancer is unique, with its own set of genetic changes and traits. Some cancers grow quickly while others can take years to become dangerous to the patient. The many differences between cases of cancer, even of the same organ (i.e. different cases of breast cancer), is one of the main reasons that treating and curing cancer is so difficult. Despite the differences between different types of cancer, all cancers DO share some common features, and these shared properties are the basis for many cancer treatments and research efforts. It is important to understand the basic, shared, features of cancer. This will allow an understanding of detection, diagnosis and treatment options. There are more than 200 different types of cancer. Worldwide, 1 in 3 people will get cancer in their life time.
_____
_____
Origin of the word cancer:
The origin of the word cancer is credited to the Greek physician Hippocrates (460-370 BC), who is considered the “Father of Medicine.” Hippocrates used the terms carcinos and carcinoma to describe non-ulcer forming and ulcer-forming tumors. In Greek, these words refer to a crab, most likely applied to the disease because the finger-like spreading projections from a cancer called to mind the shape of a crab. The Roman physician, Celsus (25 BC – 50 AD), later translated the Greek term into cancer, the Latin word for crab. Galen (130-200 AD), another Greek physician, used the word oncos (Greek for swelling) to describe tumors. Although the crab analogy of Hippocrates and Celsus is still used to describe malignant tumors, Galen’s term is now used as a part of the name for cancer specialists – oncologists.
_
World Cancer Day:
World Cancer Day is an international day marked on 4 February to raise awareness of cancer and to encourage its prevention, detection, and treatment. World Cancer Day is led by the Union for International Cancer Control (UICC) to support the goals of the World Cancer Declaration, written in 2008. The primary goal of World Cancer Day is to significantly reduce illness and death caused by cancer and is an opportunity to rally the international community to end suffering from cancer. The day is observed by the United Nations. World Cancer Day targets misinformation, raises awareness, and reduces stigma. Multiple initiatives are run on World Cancer Day to show support for those affected by cancer. One of these movements are #NoHairSelfie, a global movement to have “hairticipants” shave their heads either physically or virtually to show a symbol of courage for those undergoing cancer treatment. Images of participants are then shared all over social media. Hundreds of events around the world also take place.
_
Oncology:
The branch of medicine dedicated to diagnosing, treating and researching cancer is known as oncology, while a physician who works in the field is called an oncologist. Some oncologists focus solely on particular cancer types or treatments. Depending on the type, stage and location of a cancer, multiple oncology specialists may be involved in a patient’s care. The field of oncology has three main specialties—medical, surgical and radiation—and numerous sub-specialties. A medical oncologist is a licensed physician (typically in internal medicine) trained in diagnosing, staging and treating cancer. This specialist also leads the development of the cancer patient’s treatment plan, which may include surgery, chemotherapy, immunotherapy, targeted therapy or hormone therapy, while also coordinating with other oncology specialists and clinicians who may have a role in the patient’s care. A medical oncologist is also the doctor a cancer patient will continue to see after treatment, for check-ups over the long-term. A surgical oncologist is a surgeon who specializes in performing biopsies and removing cancerous tumors and surrounding tissue, as well as other cancer-related operations. A radiation oncologist specializes in treating cancer with radiation therapy to shrink or destroy cancer cells or to ease cancer-related symptoms. Many cancer types are treated by an oncology sub-specialty. Gynaecologic oncologists, for example, are trained to treat cancers of the female reproductive system such as those affecting the uterus, cervix, or ovaries, while hematologic oncologists specialize in diagnosing and treating blood cancers (leukemia, lymphoma and multiple myeloma). A neuro-oncologist treats cancers of the brain, spine and peripheral nerves.
_____
_____
Cancer may produce symptoms in one or more of the following ways:
- Mass effect:
An abnormal growth of tissue, or tumor, may compress nearby structures, causing pain, inflammation or disruption of function. Not all cancers produce solid tumors. Even benign cancers (those that do not metastasize, or spread to other tissues) may have serious consequences if they appear in dangerous places, particularly the heart or brain. Small bowel obstructions caused by the growth of a tumor in the digestive system is another example of a ‘space-occupying’ consequence of cancer.
- Loss of Function:
Tumor cells may deplete normal cells of oxygen and nutrients, thus disrupting the function of a vital organ. Many tumors stimulate new blood vessel formation which serves to supply the tumor rather than the normal, healthy tissue. The abnormal function of cancer cells and reduced function of normal cells in a given organ may lead to organ failure.
- Increased Lactate Production:
The Warburg Effect states that cancer cells in the presence of oxygen and glucose take a different path of energy production, diverting energy for biomass production to support tumor growth. This unique metabolism of cancer cells opens doors for possible cancer treatments including targeting lactate dehydrogenase and TCA intermediate production.
- Paraneoplastic Syndromes:
Some cancers produce “ectopic” hormones, particularly when tumors arise from neuroendocrine cells, causing a variety of endocrine imbalances. Examples include the production of parathyroid hormones by parathyroid tumors or serotonin by carcinoid tumors. In these cases, the cell types that produce these active small molecules proliferate malignantly and lose their responsiveness to negative feedback. Because hormones operate on tissues far from the site of production, paraneoplastic signs and symptoms may appear far from the tumor of origin.
-Venous Thromboembolism:
Patients with certain types of cancers are at increased risk of blood clots due to excess production of clotting factors. These clots may disrupt circulation locally or dislodge and travel to the heart, lungs, or brain, and may be fatal. Symptoms of blood clots may include pain, swelling, warmth and in late stages, numbness, particularly in the arms and legs. Some cancer treatments may further increase this risk.
- Effusions:
Cancers may stimulate fluid shifts in the body and lead to extracellular collections of fluid. Breast and lung cancer, for example, often cause pleural effusions, or a build-up of fluid in the lining of the lungs. Abdominal cancers, including ovarian and uterine cancers, may cause fluid build-up in the abdominal cavity.
_____
_____
The following types of cancer are classified according to their tissues:
Cancers are classified according to the tissue and cell type from which they arise. Cancers arising from epithelial cells are termed carcinomas; those arising from connective tissue or muscle cells are termed sarcomas. Cancers that do not fit in either of these two broad categories include the various leukemia derived from hemopoietic cells, and cancers derived from cells of the nervous system.
- Carcinoma – In epithelial tissues, such as the mucous membranes in the mouth and the gastrointestinal tract, carcinoma cancer occurs. A cancer case is estimated to be carcinoma 80 to 90% of the time, according to the National Cancer Institute.
- Leukemia – In the bone marrow, where blood cells are produced, leukemia develops.
- Lymphoma – Spleen, lymph nodes, tonsils, and thymus comprise the lymphatic system. Immune function and hormone activity are related in this system.
- Mixed types – In mixed cancers, two different types of cells can be involved, from the same category or from different categories.
- Myeloma – Originating in plasma cells that circulate as part of the blood, this type of cancer occurs predominantly in the bone marrow.
- Sarcoma – All of these substances are produced by connective tissues in skeletal structures, muscles, fat, and cartilage. Sarcomas generally occur as you age.
It is possible for a doctor to distinguish tumors of one type from those of another by their distinct appearance.
_
Each cancer has characteristics that reflect its origin. Thus, for example, the cells of an epidermal basal-cell carcinoma, derived from a keratinocyte stem cell in the skin, will generally continue to synthesize cytokeratin intermediate filaments, whereas the cells of a melanoma, derived from a pigment cell in the skin, will often (but not always) continue to make pigment granules. Cancers originating from different cell types are, in general, very different diseases. The basal-cell carcinoma, for example, is only locally invasive and rarely forms metastases, whereas the melanoma, if not removed promptly, is much more malignant and rapidly gives rise to many metastases (behavior that recalls the migratory tendencies of the normal pigment-cell precursors during development). The basal-cell carcinoma is usually easy to remove by surgery, leading to complete cure; but the malignant melanoma, once it has metastasized widely, is often impossible to eliminate and consequently fatal.
______
______
Epidemiology of cancer in brief:
At the international cancer congress held in Tokyo in 1966, Sir Alexander Haddow, the President of international union against cancer, pronounced: “We are impressed by the probability that a much higher proportion of human cancer than we had ever recently suspected—perhaps amounting to as much as 80 percent—may be due to environmental causes”. These remarks from an eminent cancer researcher are significant, because it suggests that most human cancers are preventable. The most common preventable risk factors for cancer are tobacco smoking or chewing, diet (low in fruits and vegetable and high in fatty foods, red meats, etc.), obesity, and alcohol. Cancer rate also increases with age, but age-related cancer patterns are fairly complex.
Epidemiology showing the definitive link between tobacco smoke and cancer was a noteworthy achievement in the United States, and the Surgeon General’s Report in 1964 had a significant positive effect on public health in this country. The smoking prevalence in males decreased by about 60%, while prevalence in females diminished by about 50%. As a result, lung cancer mortality and other tobacco-related diseases continue to decrease. These facts reiterate the importance of tobacco control in prevention of cancer and other diseases.
Epidemiology of skin cancer has also been enlightening. One in every three cancers diagnosed is a skin cancer, and one in every five Americans will develop skin cancer in their lifetime. Melanoma and nonmelanoma skin cancer (NMSC) are the most common types of cancer mainly in the white populations. Both types of tumors show an increasing incidence rate worldwide, but a stable or decreasing mortality rate, presumably due to earlier diagnosis and better treatments. NMSC is the most common cancer in fair-skinned individuals, which causes significant morbidity. The rising incidence rates of NMSC are believed to be triggered by a combination of increased exposure to direct UV rays or UV in sunlight, increased longevity, ozone depletion, genetics, and in a limited number of cases, immune suppression.
The most common types of cancer in males are lung cancer, prostate cancer, colorectal cancer, and stomach cancer. In females, the most common types are breast cancer, colorectal cancer, lung cancer, and cervical cancer. If skin cancer other than melanoma were included in total new cancer cases each year, it would account for around 40% of cases. In children, acute lymphoblastic leukemia and brain tumors are most common, except in Africa, where non-Hodgkin lymphoma occurs more often. In 2012, about 165,000 children under 15 years of age were diagnosed with cancer. The risk of cancer increases significantly with age, and many cancers occur more commonly in developed countries. Rates are increasing as more people live to an old age and as lifestyle changes occur in the developing world.
______
______
The problem:
Cancer is a leading cause of death worldwide, accounting for nearly 10 million deaths in 2020. The most common in 2020 (in terms of new cases of cancer) were:
- breast (2.26 million cases);
- lung (2.21 million cases);
- colon and rectum (1.93 million cases);
- prostate (1.41 million cases);
- skin (non-melanoma) (1.20 million cases); and
- stomach (1.09 million cases).
The most common causes of cancer death in 2020 were:
- lung (1.80 million deaths);
- colon and rectum (916000 deaths);
- liver (830000 deaths);
- stomach (769000 deaths); and
- breast (685000 deaths).
Each year, approximately 400,000 children develop cancer. The most common cancers vary between countries. Cervical cancer is the most common in 23 countries.
____
____
Cancer statistics and cost:
Cancer is one of the leading causes of death in the world. The International Agency for Research on Cancer (IARC) estimated 19.3 million new cancer cases (18.1 million excluding nonmelanoma skin cancer) and almost 10.0 million cancer deaths (9.9 million excluding nonmelanoma skin cancer) in 2020. Female breast cancer has surpassed lung cancer as the most commonly diagnosed cancer, with an estimated 2.3 million new cases (11.7%), followed by lung (11.4%), colorectal (10.0 %), prostate (7.3%), and stomach (5.6%) cancers. Lung cancer remained the leading cause of cancer death, with an estimated 1.8 million deaths (18%), followed by colorectal (9.4%), liver (8.3%), stomach (7.7%), and female breast (6.9%) cancers. Overall incidence was from 2-fold to 3-fold higher in transitioned versus transitioning countries for both sexes, whereas mortality varied <2-fold for men and little for women. Death rates for female breast and cervical cancers, however, were considerably higher in transitioning versus transitioned countries (15.0 vs 12.8 per 100,000 and 12.4 vs 5.2 per 100,000, respectively). The global cancer burden is expected to be 28.4 million cases in 2040, a 47% rise from 2020, with a larger increase in transitioning (64% to 95%) versus transitioned (32% to 56%) countries due to demographic changes, although this may be further exacerbated by increasing risk factors associated with globalization and a growing economy.
_
Key cancer facts 2022:
- Cancer is a leading cause of death worldwide, accounting for nearly 10 million deaths in 2020, or nearly one in six deaths.
- The most common cancers are breast, lung, colon and rectum and prostate cancers.
- Around one-third of deaths from cancer are due to tobacco use, high body mass index, alcohol consumption, low fruit and vegetable intake, and lack of physical activity.
- Cancer-causing infections, such as human papillomavirus (HPV) and hepatitis, are responsible for approximately 30% of cancer cases in low- and lower-middle-income countries.
- Many cancers can be cured if detected early and treated effectively.
_
Dramatic rise in cancer in people under 50:
A study by researchers from Brigham and Women’s Hospital reveals that the incidence of early onset cancers — including breast, colon, esophagus, kidney, liver, and pancreas — has dramatically increased around the world, with the rise beginning around 1990. In an effort to understand why many more people under 50 are being diagnosed with cancer, scientists conducted extensive analyses of available data, including information on early life exposures that might have contributed to the trend. Results are published in Nature Reviews Clinical Oncology. Possible risk factors for early onset cancer included alcohol consumption, sleep deprivation, smoking, obesity, and eating highly processed foods. Risk factors such as highly processed foods, sugary beverages, obesity, Type 2 diabetes, sedentary lifestyle, and alcohol consumption have all significantly increased since the 1950s.
_
Cost:
Cancer results in economic burden for patients, healthcare systems, and countries due to healthcare spending, and productivity losses from morbidity and premature mortality. Economic analyses can inform resource allocation decisions and investments in cancer control programs, including prevention, early detection, treatment, survivorship, and end-of-life care. The total cost of cancer to the global economy will reach 25.2 trillion international dollars between 2020 and 2050, according to an analysis of 29 cancers across 204 countries. Of that, five types of cancer will account for roughly half of that cost. The study, published in JAMA Oncology, found five cancers will account for nearly half that cost. Tracheal, bronchial and lung cancer were the most costly, followed by colon and rectal, breast, liver cancer and leukemia. Those cancers alone are expected to cost 12 trillion dollars between 2020 and 2050. Three-quarters of deaths by cancer occur in low- and middle-income countries, but more than half of the global cost of cancer will surface in high-income countries, with China and the U.S. bearing the highest burden, according to the analysis. Part of that outsized burden is due to large populations but also is attributed to high healthcare costs in the U.S. Without further investment in research and prevention, the cost of cancer will add up between healthcare costs, lost labor and spent savings, according to the study.
_____
_____
Causes in brief:
Cancer arises from the transformation of normal cells into tumour cells in a multi-stage process that generally progresses from a pre-cancerous lesion to a malignant tumour. These changes are the result of the interaction between a person’s genetic factors and three categories of external agents, including:
- physical carcinogens, such as ultraviolet and ionizing radiation;
- chemical carcinogens, such as asbestos, components of tobacco smoke, alcohol, aflatoxin (a food contaminant), and arsenic (a drinking water contaminant); and
- biological carcinogens, such as infections from certain viruses, bacteria, or parasites.
WHO, through its cancer research agency, the International Agency for Research on Cancer (IARC), maintains a classification of cancer-causing agents.
The incidence of cancer rises dramatically with age, most likely due to a build-up of risks for specific cancers that increase with age. The overall risk accumulation is combined with the tendency for cellular repair mechanisms to be less effective as a person grows older.
Risk factors:
Tobacco use, alcohol consumption, unhealthy diet, physical inactivity and air pollution are risk factors for cancer and other noncommunicable diseases. Tobacco use is the cause of about 22% of cancer deaths. Another 10% are due to obesity, poor diet, lack of physical activity or excessive drinking of alcohol. Other factors include certain infections, exposure to ionizing radiation, and environmental pollutants. In the developing world, 15% of cancers are due to infections such as Helicobacter pylori, hepatitis B, hepatitis C, human papillomavirus infection, Epstein–Barr virus and human immunodeficiency virus (HIV). These factors act, at least partly, by changing the genes of a cell. Typically, many genetic changes are required before cancer develops. Approximately 5–10% of cancers are due to inherited genetic defects.
_____
_____
Genetics and Cancerous Cells:
Is cancer a genetic disease?
Yes, cancer is a genetic disease.
It is caused by changes in genes that control the way cells grow and multiply. Cells are the building blocks of your body. Each cell has a copy of your genes, which act like an instruction manual. Genes are sections of DNA that carry instructions to make a protein or several proteins. Scientists have found hundreds of DNA and genetic changes (also called variants, mutations, or alterations) that help cancer form, grow, and spread. Genetic changes that cause cancer can be inherited or arise from certain environmental exposures. Genetic changes can also happen because of errors that occur as cells divide.
Cancer-related genetic changes can occur because:
- random mistakes in our DNA happen as our cells multiply
- our DNA is altered by carcinogens in our environment, such as chemicals in tobacco smoke, UV rays from the sun, and the human papillomavirus (HPV)
- they were inherited from one of our parents
DNA changes, whether caused by a random mistake or by a carcinogen, can happen throughout our lives and even in the womb. While most genetic changes aren’t harmful on their own, an accumulation of genetic changes over many years can turn healthy cells into cancerous cells. The vast majority of cancers occur by chance as a result of this process over time.
_
First of all, it’s important to understand that cancer cells and all of the cells of our body share roughly the same 25,000 genes. The difference between cancer cells and healthy cells is that cancer cells turn off genes that would regulate their growth, and they turn on genes that help them to grow or proliferate. Cancer involves a genetic change to the cell that results in immortality. All cancers have a foundation within genetics. Their genetic structure becomes compromised and, depending on the severity of the mutation, the cell begins to perform unnecessary and even harmful functions. In fact, the average cancerous cell contains 60 or more mutations. This is a major hurdle in identifying exactly which change resulted in the development of cancer. Genetic changes often affect proto-oncogenes, tumor suppressor genes, and genes that repair DNA. These three genes form the primary driving forces of cancer. The first two genes are involved in cellular growth and division. DNA repair genes are those that would otherwise identify a cell with damaged nuclear material, such as DNA, repairing the cell. Unfortunately, mutations may occur within these genes, resulting in cancerous cells.
Proto-oncogenes:
The genes that code for the positive cell-cycle regulators are called proto-oncogenes. Proto-oncogenes are normal genes that, when mutated, become oncogenes—genes that cause a cell to become cancerous. A large number of oncogenes have been identified in retroviruses, and all have led to the discovery of proto-oncogenes that are integral to the control of cell growth. Proto-oncogenes control the growth and division of cells by coding for proteins that form a signaling “cascade.” This cascade relays messages from the exterior of the cell to the nucleus, where a molecular apparatus called the cell cycle clock resides. At the same time, tumour suppressor genes code for a similar cascade of inhibitory signals that also converge on the cell cycle clock. The cell cycle is a four-stage process in which the cell increases in size (G1 stage), copies its DNA (S stage), prepares to divide (G2 stage), and divides (M stage). On the basis of the stimulatory and inhibitory messages it receives, the clock “decides” whether the cell should enter the cell cycle and divide. If something goes wrong with the signaling cascades—say, if a stimulatory molecule is overproduced or an inhibitory molecule is inactivated—the clock’s decision-making ability may be impaired. The cell has taken the first step toward becoming a tumour cell.
The proteins that play a role in stimulating cell division can be classified into four groups—growth factors, growth factor receptors, signal transducers, and nuclear regulatory proteins (transcription factors). For a stimulatory signal to reach the nucleus and “turn on” cell division, four main steps must occur. First, a growth factor must bind to its receptor on the cell membrane. Second, the receptor must become temporarily activated by this binding event. Third, this activation must stimulate a signal to be transmitted, or transduced, from the receptor at the cell surface to the nucleus within the cell. Finally, transcription factors within the nucleus must initiate the transcription of genes involved in cell proliferation. (Transcription is the process by which DNA is converted into RNA. Proteins are then made according to the RNA blueprint, and transcription is therefore crucial as an initial step in protein production.)
Any one of the four steps outlined above can be sabotaged by a defective proto-oncogene and lead to malignant transformation of the cell. Consider what might happen to the cell cycle in a cell with a recently acquired oncogene. In most instances, the alteration of the DNA sequence will result in a less functional (or non-functional) protein. The result is detrimental to the cell and will likely prevent the cell from completing the cell cycle; however, the organism is not harmed because the mutation will not be carried forward. If a cell cannot reproduce, the mutation is not propagated and the damage is minimal. Occasionally, however, a gene mutation causes a change that increases the activity of a positive regulator. For example, a mutation that allows Cdk, a protein involved in cell-cycle regulation, to be activated before it should be could push the cell cycle past a checkpoint before all of the required conditions are met. If the resulting daughter cells are too damaged to undertake further cell divisions, the mutation would not be propagated and no harm comes to the organism. However, if the atypical daughter cells are able to divide further, the subsequent generation of cells will likely accumulate even more mutations, some possibly in additional genes that regulate the cell cycle. The Cdk example is only one of many genes that are considered proto-oncogenes. In addition to the cell-cycle regulatory proteins, any protein that influences the cycle can be altered in such a way as to override cell-cycle checkpoints. Once a proto-oncogene has been altered such that there is an increase in the rate of the cell cycle, it is then called an oncogene. Another example of that defect can be seen in the ras family of oncogenes. The ras oncogene has a single defect in its nucleotide sequence, and, as a result, there is a change of a single amino acid in the protein for which it encodes. The ras protein is important in the signal transduction pathway; mutant proteins encoded by a mutant ras gene constantly send activation signals along the cascade, even when not stimulated to do so. Overactive ras proteins are found in about 25 percent of all human cancers, including carcinomas of the pancreas, lung, and colon.
Tumor Suppressor Genes:
Like proto-oncogenes, many of the negative cell-cycle regulatory proteins were discovered in cells that had become cancerous. Tumor suppressor genes are genes that code for the negative regulator proteins, the type of regulator that—when activated—can prevent the cell from undergoing uncontrolled division. The collective function of the best-understood tumor suppressor gene proteins, retinoblastoma protein (RB1), p53, and p21, is to put up a roadblock to cell-cycle progress until certain events are completed. A cell that carries a mutated form of a negative regulator might not be able to halt the cell cycle if there is a problem.
Mutated p53 genes have been identified in more than half of all human tumor cells. This discovery is not surprising in light of the multiple roles that the p53 protein plays at the G1 checkpoint. The p53 protein activates other genes whose products halt the cell cycle (allowing time for DNA repair), activates genes whose products participate in DNA repair, or activates genes that initiate cell death when DNA damage cannot be repaired. A damaged p53 gene can result in the cell behaving as if there are no mutations. This allows cells to divide, propagating the mutation in daughter cells and allowing the accumulation of new mutations. In addition, the damaged version of p53 found in cancer cells cannot trigger cell death. Human papillomavirus can cause cervical cancer. The virus encodes E6, a protein that binds p53.
The loss of p53 function has other repercussions for the cell cycle. Mutated p53 might lose its ability to trigger p21 production. Without adequate levels of p21, there is no effective block on Cdk activation. Essentially, without a fully functional p53, the G1 checkpoint is severely compromised and the cell proceeds directly from G1 to S regardless of internal and external conditions. At the completion of this shortened cell cycle, two daughter cells are produced that have inherited the mutated p53 gene. Given the non-optimal conditions under which the parent cell reproduced, it is likely that the daughter cells will have acquired other mutations in addition to the faulty tumor suppressor gene. Cells such as these daughter cells quickly accumulate both oncogenes and non-functional tumor suppressor genes. Again, the result is tumor growth.
_
When we talk about cancer genetics, we’re talking about these two different types of genetics that are interrelated. One is the genetics of the tumor or the cancer—what’s being turned on and what’s being turned off. The second is our constitutional genetics, or our germline genetics—the genes that we’re born with or that we’ve inherited from our parents.
Most cancers are due to mutations that occur specifically within the group of the cells or the tissues that have cancer, and are not due to an inherited, genetic, germline susceptibility. The vast majority of cancer (about 90 percent) occur by chance due to what we call “sporadic mutations,” and its only five percent to 10 percent that are due to genes that we’re born with.
Most people who have a family history of cancer are not at increased risk for developing cancers because most cancers aren’t due to an inherited susceptibility. However, there are some families where there is a clear genetic susceptibility or inherited risk for cancer. Those are families that have things like Lynch Syndrome, or families with BRCA1 mutations, BRCA2 mutations, or Li-Fraumeni Syndrome. People from those families can undertake genetic testing to help clarify their risk and determine what cancers they’re at increased risk for. If they have that genetic susceptibility to cancer, they would be screened differently. Often, those individuals are screened earlier than people in the general population; they’re screened more frequently than people in the general population; and they’re also screened with different tools than folks in the general population.
_
Hereditary, familial and sporadic cancers:
All cancer is considered to be a genetic process, as it is due to genetic changes that accumulate in the deoxyribonucleic acid (DNA) of an individual throughout their lifetime and alter the way the cells grow and divide. However, genetic changes causing an increased risk for certain cancers can also be inherited from parent to child if these changes are present in the germ cells (i.e., egg and sperm) of a parent. When an individual inherits a mutated copy of a gene related to hereditary cancer, that individual is at an increased risk throughout their lifetime for developing cancer. It is estimated that 5% to 10% of cancer is due to a specific inherited genetic cause. The majority of cancer occurs sporadically. Sporadic cancer occurs when there are genetic mutations that accumulate in the DNA of an individual over time. This occurs due to random chance, lifestyle choices, and certain environmental factors and is not due to an inherited genetic mutation. Sporadic cancer occurs due to a combination of both modifiable and nonmodifiable risk factors. Examples of nonmodifiable risk factors include aging, ethnicity, and gender; examples of modifiable risk factors include alcohol and tobacco use, physical inactivity, and occupational exposures.
_
There is a difference between familial cancers and inherited cancers. Familial cancer is used to describe a situation in which more members of a particular family are diagnosed with a type of cancer than would be statistically expected, but it’s not known why; hereditary and lifestyle causes separately or together may contribute to the high incidence in the family. In contrast, the term “inherited cancer” is used to refer to familial cancers in which a genetic cause has been identified. The children of parents with an inherited cancer genetic mutation have a 50 percent chance of also having the mutation. If you have several family members who had cancer or some family members with cancer at a relatively young age (younger than age 50) talk to your doctor about genetic testing for yourself and your family.
_
The existence of genetic predisposition to cancer is illustrated by well-defined heritable cancer syndromes. Over 200 such syndromes have now been defined. Even though they account for only 5% to 10% of all human cancers, studies of families with these syndromes provided many of the initial clues to understanding the genetic basis of sporadic (nonheritable) cancers. Although inheritance is recessive, these familial cancer syndromes show dominant patterns of inheritance and many have high penetrance. All but two of the known familial cancer syndromes are due to mutations that inactivate tumor suppressor genes. As originally proposed by Knudson in his “two-hit” hypothesis from studies in children with retinoblastoma, individuals at risk are obligate heterozygotes (they inherit a mutant allele and a wild-type allele). As it happens, homozygous mutations in critical growth regulatory genes usually cause embryonic lethality; however, in the case in which a single allele is affected, the mutation is present in every cell in the body. Given the rate of spontaneous mutation as described, the probability that the second, wild-type allele will be inactivated in at least one cell is extremely high, therefore facilitating tumorigenesis. This process is called loss of heterozygosity (LOH).
A curious observation worth noting is that different mutations in a single gene can predispose individuals to distinct cancer syndromes, whereas independent, single mutations of different genes can result in virtually the same disease, or at least diseases with indistinguishable phenotypes. This is not surprising when we consider that commonly affected genes are multifunctional and parts of complex, interactive networks or circuits as seen in the figure below. Thus a mutation may only alter gene function along one biochemical pathway, leaving its interactions with other pathways intact. Moreover, mutations that contribute to most sporadic cancers are restricted to a small subset of genes, many of which also are associated with heritable cancer syndromes.
_
Examples of inherited cancers:
Inherited cancers are those caused by a mutation in a gene that was present in the egg or sperm cell at the time of fertilization. These cancers make up a fraction of common cancers—like breast, colon, and prostate cancer—as well as less common cancers like pancreatic and ovarian cancer. It’s important to note that the presence of the mutation does not mean development of cancer is inevitable.
Below are some of the well-known genetic mutations that can be passed on to children:
- BRCA1 or BRCA2. Women and men with BRCA1 or BRCA2 genetic mutations have a significantly higher risk of developing breast and other cancers than those without the mutations. Other cancers associated with the BRCA1 and BRCA2 genes include ovarian, pancreatic, prostate, and melanoma.
- Mutations in specific mismatch repair genes that cause Lynch syndrome. This is a hereditary disorder caused by mutations in the mismatch repair genes MLH1, MSH2, MSH6, PMS2, and EPCAM. People born with mutations in these genes have a significantly higher risk of developing colon, endometrial, and other cancers such as stomach, pancreas, urinary tract, or brain.
- CDH1. Mutations in the CDH1 gene are associated with hereditary diffuse gastric cancer, a rare type of stomach cancer, as well as breast cancer.
- PALB2. Inherited mutations in the PALB2 gene are associated with an increased risk of breast cancer. Sometimes, this mutation can also be found in families with multiple cases of pancreatic cancer.
- STK11. A mutation in the STK11 gene causes Peutz-Jeghers syndrome. People with this condition have a higher-than-average risk of developing cancers of the breast, cervix, ovary, gastrointestinal tract, and pancreas.
- PTEN. Mutations in the PTEN gene have been linked to an increased risk of breast, head and neck squamous cell carcinoma, lung, and prostate cancers.
- CDKN2A or CDK4. Changes in the CDKN2A or CDK4 genes have been linked to an increased risk of melanoma.
- RB. Retinoblastomas are very rare cancers that develop in the eyes of children and have been linked to mutations in the RB gene.
- RET. A very specific type of thyroid cancer, called medullary carcinoma of the thyroid, is linked to germline mutations in the RET gene.
_
From a study of 44,788 pairs of twins, it is estimated that overall, about 30% of cancers are familial (largely due to inherited germ line mutations or genetic polymorphisms) and 70% are sporadic. From a study of 80,309 monozygotic and 123,382 same-sex dizygotic twin individuals (N = 203,691) within the population-based registers of Denmark, Finland, Norway, and Sweden, heritability of cancer overall was 33%. Significant heritability was observed for the cancer types of prostate, melanoma, breast, ovary, and uterus.
_
Cancer is a pathologic accumulation of clonally expanded cells derived from a common precursor. The fundamental cause of all cancers is genetic damage, which is usually acquired but is sometimes congenital. In general, the genetic dysregulation that gives rise to uncontrolled cell proliferation results from activation of growth- promoting oncogenes and/or deletion/inactivation of growth-inhibiting tumor suppressor genes. Additional contributions to carcinogenesis come from genes that regulate programmed cell death (apoptosis) and genes involved in DNA repair. Knudson “2-hit” hypothesis posits that a mutation in one predisposing gene is necessary but not sufficient for malignancy and that only after development of a second mutation will invasive cancer develop. Normal cell requires two successive mutations to delete both functional copies of a given gene to develop cancer. In contrast, cells with germline mutations only require a single genetic event to develop malignancy. In addition, it has become increasingly apparent that cancer cells must evade host immune responses and this can sometimes be exploited to treat cancer.
_____
_____
Basics of cancer:
The molecular basis of cancer:
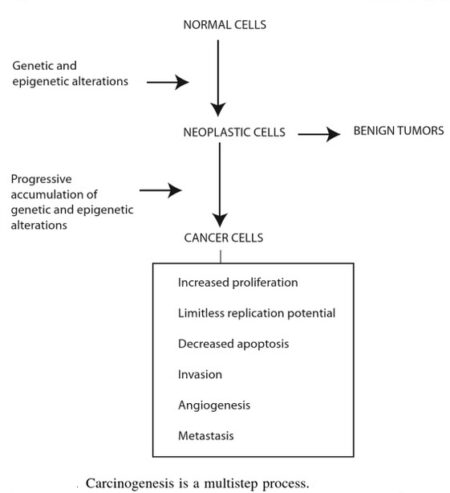
Discussion of the causes of cancers necessarily involves an examination of the molecular machinery in cells that guides the basic processes of proliferation (increase in cell number by cell division), differentiation (cell specialization into different tissue types), and apoptosis (programmed cell death). Those processes are guided by two innate programs in cells, the genetic code and the epigenetic code. In cancer each of those codes ultimately becomes altered regardless of whether the disease originated with an external or internal factor. Indeed, a fundamental characteristic of a tumour cell is that it begets a tumour cell. In other words, cancer, once manifest, becomes an inherited disease of the cell and is therefore self-perpetuating. The hereditary nature of cancer at the cellular level explains why alterations have been found in both the genetic and the epigenetic codes in tumour cells. The number of alterations seen in the coded programs increases as tumours progress to more advanced stages. Their existence and accumulation also explain why principles of evolutionary theory provide insights of practical significance for cancer biology.
_
_
Genetic and epigenetic programs:
One way to envision a cancer cell is to think of a cell that has rewired the normal control circuits for proliferation, differentiation, and death. The resulting alterations in the circuits’ functions, which are encoded by the genetic sequence and by the epigenetic configuration, enable the cell to escape programmed controls.
The genetic program, common to all cells in the body (whether noncancerous or cancerous), is found in the DNA sequence, which is packaged in chromosomes in the cell nucleus. Each person has a unique DNA sequence that is composed of approximately three billion base pairs (units of DNA) per haploid set of chromosomes organized into roughly 25,000 genes. A gene can be thought of as a set of instructions that the cell follows to make a protein, each gene providing directions for a different protein. Some of the gene products that have been linked to cancer are organized in groups (pathways), which form networks that transmit information inside the cell and stimulate responses to changes in the cell’s environment.
The epigenetic code is responsible for providing cells with the memory of their particular specialization—for example, being part of the brain, the liver, or skin. The epigenetic code is embodied in chemical changes to DNA and in chemical and structural modifications of chromatin (the protein-DNA fibres in the nucleus that when condensed form the chromosomes). When located in a gene promoter, DNA methylation typically acts to repress gene transcription.
The billions of cells that make up a tumour are descended from a single cell, in which disturbance of the genetic and epigenetic codes caused remodeling of the control circuits that governed that cell’s existence. A single damaging genetic or epigenetic event, however, is not enough to convert a healthy cell to a cancer cell. Rather, several insults must be inflicted upon the DNA or chromatin of a cell in order for it to become cancerous. The first of those, the damage that instigates transformation, is known as initiation. Ensuing damage that advances transformation is known as promotion. Initiation and promotion together are required for causing cancer. In many cases that is a slow process that takes years.
_
Cancer cells are created when the genes responsible for regulating cell division are damaged. Carcinogenesis is caused by mutation and epimutation of the genetic material of normal cells, which upsets the normal balance between proliferation and cell death. This results in uncontrolled cell division in the body. The uncontrolled and often rapid proliferation of cells can lead to benign or malignant tumours (cancer). Benign tumors do not spread to other parts of the body or invade other tissues. Malignant tumors can invade other organs, spread to distant locations (metastasis) and become life-threatening. More than one mutation is necessary for carcinogenesis. In fact, a series of several mutations to certain classes of genes is usually required before a normal cell will transform into a cancer cell. Damage to DNA can be caused by exposure to radiation, chemicals, and other environmental sources, but mutations also accumulate naturally over time through uncorrected errors in DNA transcription, making age another risk factor. Oncoviruses can cause certain types of cancer, and genetics are also known to play a role.
_
Cancer begins when a cell breaks free from the normal restraints on cell division and begins to follow its own agenda for proliferation (Figure below). All of the cells produced by division of this first, ancestral cell and its progeny also display inappropriate proliferation. A tumor, or mass of cells, formed of these abnormal cells may remain within the tissue in which it originated (a condition called in situ cancer), or it may begin to invade nearby tissues (a condition called invasive cancer). An invasive tumor is said to be malignant, and cells shed into the blood or lymph from a malignant tumor are likely to establish new tumors (metastases) throughout the body. Tumors threaten an individual’s life when their growth disrupts the tissues and organs needed for survival.
Figure below shows stages of tumor development.
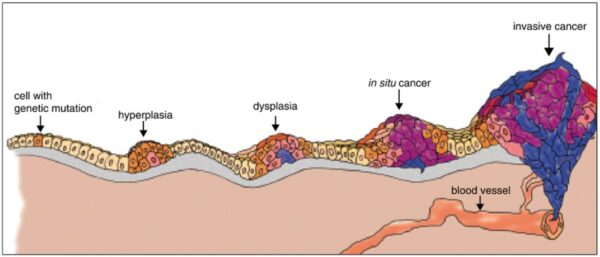
A malignant tumor develops across time, as shown in above diagram. This tumor develops as a result of four mutations, but the number of mutations involved in other types of tumors can vary. We do not know the exact number of mutations required for a normal cell to become a fully malignant cell, but the number is probably less than ten.
-a. The tumor begins to develop when a cell experiences a mutation that makes the cell more likely to divide than it normally would.
-b. The altered cell and its descendants grow and divide too often, a condition called hyperplasia. At some point, one of these cells experiences another mutation that further increases its tendency to divide.
-c. This cell’s descendants divide excessively and look abnormal, a condition called dysplasia. As time passes, one of the cells experiences yet another mutation.
-d. This cell and its descendants are very abnormal in both growth and appearance. If the tumor that has formed from these cells is still contained within its tissue of origin, it is called in situ cancer. In situ cancer may remain contained indefinitely.
-e. If some cells experience additional mutations that allow the tumor to invade neighboring tissues and shed cells into the blood or lymph, the tumor is said to be malignant. The escaped cells may establish new tumors (metastases) at other locations in the body.
_
What happens to cause a cell to become cancerous?
Thirty years ago, scientists could not offer a coherent answer to this question. They knew that cancer arose from cells that began to proliferate uncontrollably within the body, and they knew that chemicals, radiation, and viruses could trigger this change. But exactly how it happened was a mystery.
Research across the last three decades, however, has revolutionized our understanding of cancer. In large part, this success was made possible by the development and application of the techniques of molecular biology, techniques that enabled researchers to probe and describe features of individual cells in ways unimaginable a century ago. Today, we know that cancer is a disease of molecules and genes, and we even know many of the molecules and genes involved. In fact, our increasing understanding of these genes is making possible the development of exciting new strategies for avoiding, forestalling, and even correcting the changes that lead to cancer.
_
Cancer as a Multistep Process:
A central feature of today’s molecular view of cancer is that cancer does not develop all at once, but across time, as a long and complex succession of genetic changes. Each change enables precancerous cells to acquire some of the traits that together create the malignant growth of cancer cells.
Two categories of genes play major roles in triggering cancer. In their normal forms, these genes control the cell cycle, the sequence of events by which cells enlarge and divide. One category of genes, called proto-oncogenes, encourages cell division. The other category, called tumor suppressor genes, inhibits it. Together, proto-oncogenes and tumor suppressor genes coordinate the regulated growth that normally ensures that each tissue and organ in the body maintains a size and structure that meets the body’s needs.
What happens when proto-oncogenes or tumor suppressor genes are mutated? Mutated proto-oncogenes become oncogenes, genes that stimulate excessive division. And mutations in tumor suppressor genes inactivate these genes, eliminating the critical inhibition of cell division that normally prevents excessive growth. Collectively, mutations in these two categories of genes account for much of the uncontrolled cell division that occurs in human cancers.
_
The body’s back-up systems:
In addition to the controls on proliferation afforded by the coordinated action of proto-oncogenes and tumor suppressor genes, cells also have at least three other systems that can help them avoid runaway cell division.
-1. The first of these systems is the DNA repair system. This system operates in virtually every cell in the body, detecting and correcting errors in DNA. Across a lifetime, a person’s genes are under constant attack, both by carcinogens imported from the environment and by chemicals produced in the cell itself. Errors also occur during DNA replication. In most cases, such errors are rapidly corrected by the cell’s DNA repair system. Should the system fail, however, the error (now a mutation) becomes a permanent feature in that cell and in all of its descendants. The system’s normally high efficiency is one reason why many years typically must pass before all the mutations required for cancer to develop occur together in one cell. Mutations in DNA repair genes themselves, however, can undermine this repair system in a particularly devastating way: They damage a cell’s ability to repair errors in its DNA. As a result, mutations appear in the cell (including mutations in genes that control cell growth) much more frequently than normal.
-2. A second cellular back-up system prompts a cell to commit suicide (undergo apoptosis) if some essential component is damaged or its control system is deregulated. This observation suggests that tumors arise from cells that have managed to evade such death. One way of avoiding apoptosis involves the p53 protein. In its normal form, this protein not only halts cell division, but induces apoptosis in abnormal cells. The product of a tumor suppressor gene, p53 is inactivated in many types of cancers. This ability to avoid apoptosis endangers cancer patients in two ways. First, it contributes to the growth of tumors. Second, it makes cancer cells resistant to treatment.
-3. A third back-up system limits the number of times a cell can divide, and so assures that cells cannot reproduce endlessly. This system is governed by a counting mechanism that involves the DNA segments at the ends of chromosomes. Called telomeres, these segments shorten each time a chromo some replicates. Once the telomeres are shorter than some threshold length, they trigger an internal signal that causes the cell to stop dividing. If the cells continue dividing, further shortening of the telomeres eventually causes the chromosomes to break apart or fuse with one another, a genetic crisis that is inevitably fatal to the cell. Early observations of cancer cells grown in culture revealed that, unlike normal cells, cancer cells can proliferate indefinitely. Scientists have recently discovered the molecular basis for this characteristic—an enzyme called telomerase, that systematically replaces telomeric segments that are trimmed away during each round of cell division. Telomerase is virtually absent from most mature cells, but is present in most cancer cells, where its action enables the cells to proliferate endlessly.
_
The multistep development of cancer:
Cancer, then, does not develop all at once as a massive shift in cellular functions that results from a mutation in one or two wayward genes. Instead, it develops step-by-step, across time, as an accumulation of many molecular changes, each contributing some of the characteristics that eventually produce the malignant state. The number of cell divisions that occur during this process can be astronomically large—human tumors often become apparent only after they have grown to a size of 10 billion to 100 billion cells. As you might expect, the time frame involved also is very long— it normally takes decades to accumulate enough mutations to reach a malignant state.
Understanding cancer as a multistep process that occurs across long periods of time explains a number of long-standing observations. A key observation is the increase in incidence with age. Cancer is, for the most part, a disease of people who have lived long enough to have experienced a complex and extended succession of events. Because each change is a rare accident requiring years to occur, the whole process takes a very long time, and most of us die from other causes before it is complete.
Understanding cancer in this way also explains the increase in cancer incidence in people who experience unusual exposure to carcinogens, as well as the increased cancer risk of people who inherit predisposing mutations. Exposure to carcinogens increases the likelihood that certain harmful changes will occur, greatly increasing the probability of developing cancer during a normal life span. Similarly, inheriting a cancer -susceptibility mutation means that instead of that mutation being a rare event, it already has occurred, and not just in one or two cells, but in all the body’s cells. In other words, the process of tumor formation has leapfrogged over one of its early steps. Now the accumulation of changes required to reach the malignant state, which usually requires several decades to occur, may take place in one or two.
Finally, understanding the development of cancer as a multistep process also explains the lag time that often separates exposure to a cancer-causing agent and the development of cancer. This explains, for example, the observation that severe sunburns in children can lead to the development of skin cancer decades later. It also explains the 20-to 25-year lag between the onset of widespread cigarette smoking among women after World War II and the massive increase in lung cancer that occurred among women in the 1970s.
_
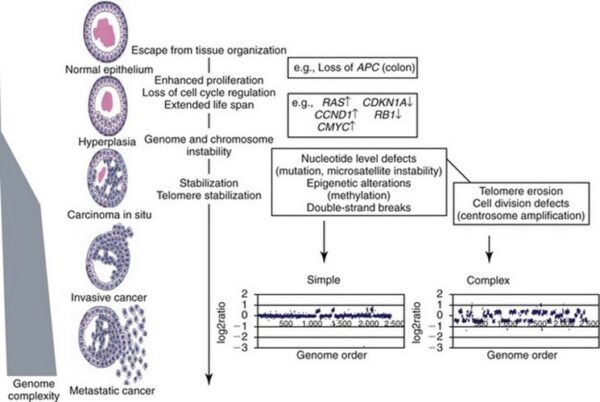
Figure above shows schematic representation of chromosomal evolution in human solid tumor progression. The stages of progression are arranged with the earlier lesions at the top. Cells may begin to proliferate excessively owing to the loss of tissue architecture, abrogation of checkpoints, and other factors. In general, relatively few aberrations occur before the development of in situ cancer. As indicated, a sharp increase in genome complexity (the number of independent chromosomal aberrations) in many (but not all) tumors coincides with the development of in situ disease. The types and range in aberration number varies markedly between tumors, probably owing to the specific failures in checkpoint or damage surveillance that are present, as illustrated by the whole-genome array comparative genomic hybridization (CGH) profiles of HCT116, a mismatch repair-defective cell line, and T47D, a mismatch repair-proficient cell line. The copy number profiles of HCT116 and T47D are labeled as “simple” and “complex,” respectively, to distinguish between tumor genomes with few or many copy number changes. The spectrum of aberrations in in situ lesions is similar to those found in more advanced malignancies. Thus an early increase in chromosomal aberration composition is followed by more modest chromosomal evolution.
_____
_____
Precancerous conditions:
A precancerous condition is a condition, tumor or lesion involving abnormal cells which are associated with an increased risk of developing into cancer. Clinically, precancerous conditions encompass a variety of abnormal tissues with an increased risk of developing into cancer. Some of the most common precancerous conditions include certain colon polyps, which can progress into colon cancer, monoclonal gammopathy of undetermined significance, which can progress into multiple myeloma or myelodysplastic syndrome. and cervical dysplasia, which can progress into cervical cancer. Bronchial premalignant lesions can progress to squamous cell carcinoma of the lung.
_
Precancerous cells are also called premalignant cells. They are defined as abnormal cells that could turn into cancerous cells, but which, by themselves, are not invasive or spreading. The concept of precancerous cells, and whether they progress or not, may sometimes be confusing. That’s because the answer isn’t always certain. In general, cells don’t go from normal on day one, to premalignant on day two, and then on to cancer on day three. Sometimes precancerous cells progress to cancer, but more often they don’t. They may stay the same—that is, remain abnormal but not invasive—or they may even become normal again.
Again, it’s important to note that cells that are precancerous are not cancer cells. This means that left alone, they will not spread to other regions of the body. They are simply abnormal cells that could, in time, undergo changes that would transform them into cancer cells. If the cells are removed before they become cancer cells, then the condition should, in theory, be 100% curable. That said, not all precancerous cells need to be removed right away.
Precancerous conditions are often identified through routine screenings such as blood tests that are part of a standard physical exam. Other times, they’re found when an individual has a specific symptom or health issue. Screening for precancerous conditions has become a routine part of medical examinations, especially as individuals age and become more susceptible to certain types of cancer. Pap tests, for example, which involve collecting cells from the cervix and examining them for abnormalities, can help identify women at risk for cervical cancer and are generally recommended for women beginning at age 21. In parts of the world where the Pap test is widely used, cervical cancer rates have dropped sharply.
One point of confusion is that cancer cells and precancerous cells occur together in many tumors. For example, some people with breast cancer have cancer cells in a tumor, but there may be other regions in the breasts and even in the tumor itself in which precancerous cells are found as well.
_
Pathologically, precancerous tissue can range from benign neoplasia, which are tumors which don’t invade neighboring normal tissues or spread to distant organs, to dysplasia, a collection of highly abnormal cells which, in some cases, has an increased risk of progressing to anaplasia and invasive cancer which is life-threatening. The word “dysplasia” is often used to mean the same thing as “precancerous cells,” yet there are a few differences. In many cases, when healthcare providers speak of dysplasia, they are indeed talking about these abnormal cells that could turn into cancer cells. But in some cases, the term “severe dysplasia” is used to describe cells that are already cancerous. They are still contained within the tissues in which they began and have not spread. This is known as carcinoma in situ, which is a non-invasive cancer that has not grown and spread to nearby tissue, unlike the invasive stage. As with other precancerous conditions, not all carcinoma in situ will become an invasive disease but is at risk of. Precancerous changes are usually described in degrees or levels of abnormality. Severity and grade are the two main ways that are used to describe them.
_
Types of Precancerous Conditions:
Cancers that begin in epithelial cells (roughly 85% of cancers) may have a precancerous state before they turn into cancer. These cells are found in the skin and the lining tissue of many organs.
Some precancerous conditions include:
- Cervical intraepithelial neoplasia (CIN): A precancerous state of cervical cancer
- Barrett’s esophagus: Abnormal cells that may go on to become esophageal cancer
- Atypical lobular hyperplasia: May develop into breast cancer
- Adenomatous polyps in the colon: May develop into colon cancer
- Actinic keratoses: Abnormal changes in the skin that may develop into squamous cell skin cancer
- Dysplastic moles: May develop into melanoma or indicate a higher risk of melanoma
- Bronchial epithelial dysplasia: May develop into lung cancer
- Atrophic gastritis: Abnormal changes in the stomach that may develop into gastric (stomach) cancer
- Bowen’s disease: May develop into invasive skin cancer
_
Latency and Progression:
A discussion of precancerous cell changes offers a good opportunity to talk about another concept that can be hard to grasp. This is called latency. The latency period is defined as the period of time between exposure to a cancer-causing substance (a carcinogen) and the later development of cancer.
People are often surprised when they develop cancer many years after exposure to a carcinogen. For example, some people are diagnosed with lung cancer even when they quit smoking three decades earlier. Genetic damage is done when cells are first exposed to a cancer-causing agent. But it’s usually an accumulation of this damage, and the genetic mutations involved over time, that causes a cell to become precancerous. The cell may then progress through stages of mild to moderate—and on to severe—dysplasia before finally becoming a cancer cell. This progress toward cancer also may be limited by other factors in its environment, or it may even revert back into a normal cell. That’s why a healthy diet and exercise are important even if you’ve been exposed to a carcinogen. The process is much more complex than once thought, but understanding the basics does help to explain the latency period seen with many cancers.
When do cells become cancerous from precancerous state?
Most of the time, the answer to how long it may take for a precancerous cell to become cancerous will vary. The answer also depends on the type of cell that’s involved. In one study that looked at 101 people with abnormal cell changes of the vocal cords, 15 of them went on to develop invasive cancer. One of those had mild dysplasia, one had moderate dysplasia, seven had severe dysplasia, and six had carcinoma in situ. In 73% of these people, their precancerous lesions became invasive cancer of the vocal cords within one year. The rest of them developed cancer years later.
______
______
Theories of oncogenesis:
There are a number of theories of carcinogenesis and cancer treatment that fall outside the mainstream of scientific opinion, due to lack of scientific rationale, logic, or evidence base. These theories may be used to justify various alternative cancer treatments. They should be distinguished from those theories of carcinogenesis that have a logical basis within mainstream cancer biology, and from which conventionally testable hypotheses can be made. Several alternative theories of carcinogenesis, however, are based on scientific evidence and are increasingly being acknowledged. Some researchers believe that cancer may be caused by aneuploidy (numerical and structural abnormalities in chromosomes) rather than by mutations or epimutations. Cancer has also been considered as a metabolic disease, in which the cellular metabolism of oxygen is diverted from the pathway that generates energy (oxidative phosphorylation) to the pathway that generates reactive oxygen species. This causes an energy switch from oxidative phosphorylation to aerobic glycolysis (Warburg’s hypothesis), and the accumulation of reactive oxygen species leading to oxidative stress (“oxidative stress theory of cancer”). Another concept of cancer development is based on exposure to weak magnetic and electromagnetic fields and their effects on oxidative stress, known as magnetocarcinogenesis.
_
A number of authors have questioned the assumption that cancers result from sequential random mutations as oversimplistic, suggesting instead that cancer results from a failure of the body to inhibit an innate, programmed proliferative tendency. A related theory suggests that cancer is an atavism, an evolutionary throwback to an earlier form of multicellular life. The genes responsible for uncontrolled cell growth and cooperation between cancer cells are very similar to those that enabled the first multicellular life forms to group together and flourish. These genes still exist within the genomes of more complex metazoans, such as humans, although more recently evolved genes keep them in check. When the newer controlling genes fail for whatever reason, the cell can revert to its more primitive programming and reproduce out of control. The theory is an alternative to the notion that cancers begin with rogue cells that undergo evolution within the body. Instead, they possess a fixed number of primitive genes that are progressively activated, giving them finite variability. Another evolutionary theory puts the roots of cancer back to the origin of the eukaryote (nucleated) cell by massive horizontal gene transfer, when the genomes of infecting viruses were cleaved (and thereby attenuated) by the host, but their fragments integrated into the host genome as immune protection. Cancer thus originates when a rare somatic mutation recombines such fragments into a functional driver of cell proliferation.
_
Cancer results from uncontrolled proliferation of aberrant cells in the body. However, the mechanisms of cancer development have not been fully understood. Early studies have shown that cancer development is a multistep process. In this hypothesis, normal cells gradually become malignant through a progressive series of alterations resulting from the actions of two or more oncogenes due to gene mutations. The multistep nature of cancer development has been evidenced in prostate cancer, colorectal cancer, breast cancer, acute lymphoblastic leukemia (ALL), acute myelogenous leukemia (AML), and myeloproliferative disorders. Generally, the multistep of cancer development includes initiation, promotion, and progression. The multistep hypothesis of cancer development has an implication in cancer treatment in that the early neoplastic cells may be effectively interrupted before the malignant cells come out.
_
In 1976, Nowell proposed clonal evolution hypothesis to explain cancer development. He proposed that cancer develops by a repetitive process of clonal expansion and clonal selection within the tissue ecosystems in human body. He also proposed that clonal evolution hypothesis may have great implications in cancer treatment as cancer treatment may destroy cancer clones and corrode cancer habitats. However, it may unwittingly produce selection pressure for the clonal expansion of resistant cancer cells. For instance, there are numerous genetic subclones of leukemia-initiating cells in ALL patients. Analysis of AML patients’ samples showed relapsed AML after chemotherapy, demonstrating the generation and presence of a primary clone. Furthermore, analysis of paired persistent AML samples revealed cancer patients with 2 concomitant dominant clones; one was chemotherapy sensitive and another resistant. There is a report demonstrating that recurrent high-grade serous carcinoma (HGSC, a form of ovarian cancer) also originates from multiple clones in the primary tumor during standard treatment.
_
Cancer stem cell (CSC) hypothesis, on the other hand, was initially proposed to explain the observation of tumor heterogeneity. Also, CSCs may cause the generation of tumors in the same way as that of normal stem cells. Moreover, CSCs are capable of self-renewal and differentiation into multiple cell types in tumors, persist in tumors, and cause relapse and metastasis. Early studies indicated that CSCs might play a role in breast cancer development. It was demonstrated that CD34+CD38− cells were CSCs of leukemia, which could initiate tumors in NOD/SCID mice. At present, CSCs have been identified in many types of cancers such as brain cancer, breast cancer, colon cancer, liver cancer, lung cancer, ovary cancer, prostate cancer, pancreas cancer, gastric carcinogenesis, leukemia, melanoma, and oral carcinoma. Cancer stem cell hypothesis has great implications in the identification of cancers, selection of cancer drug targets, prevention of cancer metastasis, and development of new treatment modalities.
________
The development of cancer by mutation and natural selection among somatic cells portray microevolutionary process:
This process occurs on a time scale of months or years in a population of cells in the body, but it depends on the same principles of mutation and natural selection that govern the long-term evolution of all living organisms. Cancer cells, by definition, proliferate in defiance of normal controls (that is, they are neoplastic) and are able to invade and colonize surrounding tissues (that is, they are malignant). By giving rise to secondary tumors, or metastases, they become difficult to eradicate surgically. Most cancers are thought to originate from a single cell that has experienced an initial mutation, but the progeny of this cell must undergo further changes, requiring numerous additional mutations, to become cancerous. This phenomenon of tumor progression, which usually takes many years, reflects the unfortunate operation of evolution by mutation and natural selection among somatic cells.
_
Ultimately, all somatic cell lineages are committed to die: they leave no progeny and instead dedicate their existence to support of the germ cells, which alone have a chance of survival. There is no mystery in this, for the body is a clone, and the genome of the somatic cells is the same as that of the germ cells. By their self-sacrifice for the sake of the germ cells, the somatic cells help to propagate copies of their own genes. Thus, unlike free-living cells such as bacteria, which compete to survive, the cells of a multicellular organism are committed to collaboration. To coordinate their behavior, the cells send, receive, and interpret an elaborate set of signals that serve as social controls, telling each of them how to act. As a result, each cell behaves in socially responsible manner, resting, dividing, differentiating, or dying as needed for the good of the organism. Molecular disturbances that upset this harmony mean trouble for a multicellular society. In a human body with more than 1014 cells, billions of cells experience mutations every day, potentially disrupting the social controls. Most dangerously, a mutation may give one cell a selective advantage, allowing it to divide more vigorously than its neighbors and to become a founder of a growing mutant clone. A mutation that gives rise to such selfish behavior by individual members of the cooperative can jeopardize the future of the whole enterprise. Repeated rounds of mutation, competition, and natural selection operating within the population of somatic cells cause matters to go from bad to worse. These are the basic ingredients of cancer: it is a disease in which individual mutant clones of cells begin by prospering at the expense of their neighbors, but in the end destroy the whole cellular society.
_
Cancer cells are defined by two heritable properties: they and their progeny (1) reproduce in defiance of the normal restraints on cell division and (2) invade and colonize territories normally reserved for other cells. It is the combination of these actions that makes cancers peculiarly dangerous. An isolated abnormal cell that does not proliferate more than its normal neighbors does no significant damage, no matter what other disagreeable properties it may have; but if its proliferation is out of control, it will give rise to a tumor, or neoplasm—a relentlessly growing mass of abnormal cells. As long as the neoplastic cells remain clustered together in a single mass, however, the tumor is said to be benign. At this stage, a complete cure can usually be achieved by removing the mass surgically. A tumor is considered a cancer only if it is malignant, that is, only if its cells have acquired the ability to invade surrounding tissue. Invasiveness usually implies an ability to break loose, enter the bloodstream or lymphatic vessels, and form secondary tumors, called metastases, at other sites in the body. The more widely a cancer spreads, the harder it becomes to eradicate.
_
Most cancers derive from a Single Abnormal Cell:
Even when a cancer has metastasized, its origins can usually be traced to a single primary tumor, arising in an identified organ and presumed to be derived by cell division from a single cell that has undergone some heritable change that enables it to outgrow its neighbors. By the time it is first detected, however, a typical tumor already contains about a billion cells or more (figure below), often including many normal cells—fibroblasts, for example, in the supporting connective tissue that is associated with a carcinoma.
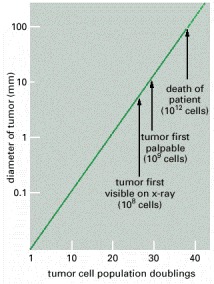
Figure above shows growth of a typical human tumor such as a tumor of the breast. The diameter of the tumor is plotted on a logarithmic scale. Years may elapse before the tumor becomes noticeable. The doubling time of a typical breast tumor, for example, is about 100 days.
_
What evidence do we have that the cancer cells are indeed a clone descended from a single abnormal cell?
One clear demonstration of clonal evolution comes from analysis of the chromosomes in tumor cells. Chromosomal aberrations and rearrangements are present in the cells of most common cancers. In almost all patients with chronic myelogenous leukemia, for example, leukemic white blood cells can be distinguished from normal cells by a specific chromosomal abnormality: the so-called Philadelphia chromosome, created by a translocation between the long arms of chromosomes 9 and 22. When the DNA at the site of translocation is cloned and sequenced, it is found that the site of breakage and rejoining of the translocated fragments is identical in all the leukemic cells in any given patient, but differs slightly (by a few hundred or thousand base pairs) from one patient to another, as expected if each case of the leukemia arises from a unique accident occurring in a single cell. Many other lines of evidence, from a variety of cancers, point to the same conclusion: most cancers originate from a single aberrant cell.
_
Cancers result from Somatic Mutations:
If a single abnormal cell is to give rise to a tumor, it must pass on its abnormality to its progeny: the aberration has to be heritable. A first problem in understanding a cancer is to discover whether the heritable aberration is due to a genetic change—that is, an alteration in the cell’s DNA sequence—or to an epigenetic change—that is, a change in the pattern of gene expression without a change in the DNA sequence. Heritable epigenetic changes, reflecting cell memory, occur during normal development, as manifest in the stability of the differentiated state and in such phenomena as X-chromosome inactivation and imprinting. Such epigenetic changes have also been found to play a part in the development of many cancers.
There are, however, good reasons to think that the vast majority of cancers are initiated by genetic changes. First, cells of a variety of cancers can be shown to have a shared abnormality in their DNA sequence that distinguishes them from the normal cells surrounding the tumor, as in the example of chronic myelogenous leukemia that we have just described.
Second, many of the agents known to give rise to cancer also cause genetic changes. Thus carcinogenesis (the generation of cancer) appears to be linked with mutagenesis (the production of a change in the DNA sequence). This correlation is clear for three classes of agents: chemical carcinogens (which typically cause simple local changes in the nucleotide sequence), ionizing radiations such as x-rays (which typically cause chromosome breaks and translocations), and viruses (which introduce foreign DNA into the cell).
Finally, the conclusion that somatic mutations underlie cancer is supported by studies of people who inherit a strong susceptibility to the disease. In a significant proportion of cases, the propensity to cancer can be traced to a genetic defect of some sort in the DNA repair mechanisms of these individuals, which allows them to accumulate mutations at an elevated rate. People with the disease xeroderma pigmentosum, for example, have defects in the cellular system that repairs DNA damage induced by UV light, and they experience a hugely increased incidence of skin cancers.
_
- A Single Mutation is not enough to cause Cancer:
An estimated 1016 cell divisions take place in a normal human body in the course of a lifetime; in a mouse, with its smaller number of cells and its shorter life span, the number is about 1012. Even in an environment that is free of mutagens, mutations will occur spontaneously at an estimated rate of about 10-6 mutations per gene per cell division—a value set by fundamental limitations on the accuracy of DNA replication and repair. Thus, in a lifetime, every single gene is likely to have undergone mutation on about 1010 separate occasions in any individual human being, or about 106 occasions in a mouse. Among the resulting mutant cells, one might expect that there would be many that have disturbances in genes that regulate cell division and that consequently disobey the normal restrictions on cell proliferation. From this point of view, the problem of cancer seems to be not why it occurs but why it occurs so infrequently.
Clearly, if a single mutation were enough to convert a typical healthy cell into a cancer cell that proliferates without restraint, we would not be viable organisms. Many types of evidence indicate that the genesis of a cancer typically requires that several independent, rare accidents occur in the lineage of one cell. One such indication comes from epidemiological studies of the incidence of cancer as a function of age. If a single mutation were responsible, occurring with a fixed probability per year, the chance of developing cancer in any given year should be independent of age. In fact, for most types of cancer the incidence rises steeply with age—as would be expected if cancer is caused by a slow accumulation of numerous random mutations in a single line of cells.
Now that many of the specific mutations responsible for the development of cancer have been identified, we can test directly for their presence in a particular case of the disease. Such tests have revealed that an individual malignant cell generally harbours multiple mutations. Animal models also confirm that a single one of these genetic alterations is insufficient to cause cancer: when genetically engineered in a mouse, a single such mutation typically produces mild abnormalities in tissue growth, followed occasionally by the formation of randomly scattered benign tumors; but the vast majority of cells in the mutant animal remain non-cancerous.
The concept that the development of a cancer requires mutations in many genes—perhaps ten or more—fits with a large body of information, dating back over many years, concerning the phenomenon of tumor progression, whereby an initial mild disorder of cell behavior evolves gradually into a full-blown cancer.
_
Tumor progression involves successive rounds of Mutation and Natural Selection:
Cancers in general seem to arise by a process in which an initial population of slightly abnormal cells, descendants of a single mutant ancestor, evolves from bad to worse through successive cycles of mutation and natural selection. At each stage, one cell acquires an additional mutation that gives it a selective advantage over its neighbors, making it better able to thrive in its environment—an environment that, inside a tumor, may be harsh, with low levels of oxygen, scarce nutrients, and the natural barriers to growth presented by the surrounding normal tissues. The offspring of this well-adapted cell will continue to divide, eventually taking over the tumor and becoming the dominant clone in the developing lesion (figure below). Thus, tumors grow in fits and starts, as additional advantageous mutations arise and the cells bearing them flourish. Their evolution involves a large element of chance and usually takes many years; most of us die of other ailments before cancer has had time to develop.
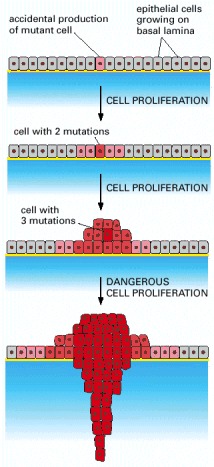
Figure above shows clonal evolution.
A tumor develops through repeated rounds of mutation and proliferation, giving rise eventually to a clone of fully malignant cancer cells. At each step, a single cell undergoes a mutation that enhances cell proliferation, so that its progeny become the dominant clone in the tumor. Proliferation of this clone then hastens occurrence of the next step of tumor progression by increasing the size of the cell population at risk of undergoing an additional mutation.
_
Why are so many mutations needed?
One reason is that cellular processes are controlled in complex and interconnected ways; cells employ redundant regulatory mechanisms to help them maintain tight and precise control over their behavior. Thus, many different regulatory systems have to be disrupted before a cell can throw off its normal restraints and behave defiantly as a malignant cancer cell. In addition, tumor cells may meet new barriers to further expansion at each stage of the evolutionary process. For example, oxygen and nutrients do not become limiting until a tumor is one or two millimeters in diameter, at which point the cells in the tumor interior may not have adequate access to such necessary resources. Each new barrier, whether physical or physiological, must be overcome by the acquisition of additional mutations.
_
In general, the rate of evolution in any population would be expected to depend on four main parameters: (1) the mutation rate, that is, the probability per gene per unit time that any given member of the population will undergo genetic change; (2) the number of individuals in the population; (3) the rate of reproduction, that is, the average number of generations of progeny produced per unit time; and (4) the selective advantage enjoyed by successful mutant individuals, that is, the ratio of the number of surviving fertile progeny they produce per unit time to the number of surviving fertile progeny produced by nonmutant individuals. These are the critical factors for the evolution of cancer cells in a multicellular organism, just as they are for the evolution of organisms on the surface of the Earth. Clearly the rate of progression toward cancer depends on many things beside the changing genotype of the individual cancer cell. Equally, it is plain that there are a number of quite disparate genetic properties that might help a cancer cell to be evolutionarily successful.
_
Most Human Cancer Cells are Genetically Unstable:
The great majority of human cancers show signs of a dramatically enhanced mutation rate: the cells are said to be genetically unstable. This instability can take various forms. Some cancer cells are defective in the ability to repair local DNA damage or to correct replication errors that affect individual nucleotides. These cells tend to accumulate more point mutations and small, localized DNA sequence changes than do normal cells. Other cancer cells have trouble maintaining the integrity of their chromosomes and thus display gross abnormalities in their karyotype. From an evolutionary perspective this is not a surprise: one might expect that genetic instability would promote the formation of cancer, as it increases the probability that cells will experience a mutation that will lead toward malignancy. In fact, it seems that some degree of genetic instability may be essential for the development of cancer; at the very least it appears to make a powerful contribution to cancer progression.
Different tumors—even from the same tissues—can show different kinds of genetic instability, caused by mutations in one or another of a very specific set of genes whose products are needed to protect the genome from alteration. People who inherit mutations in these genes are found to have a raised incidence of cancer. Although such cancer-prone conditions are relatively rare, they include examples of mutations in practically every type of gene known to be required for genetic stability, confirming that loss of this stability can have a causative role in cancer no matter how it occurs.
Most often, the destabilizing mutations are not inherited, but arise de novo as a tumor develops, helping the cancer cell to accumulate mutations much more rapidly than its neighbors do. It seems that some “optimum” level of genetic instability exists for the development of cancer, making a cell mutable enough to evolve dangerously, but not so mutable that it dies as seen in figure below.
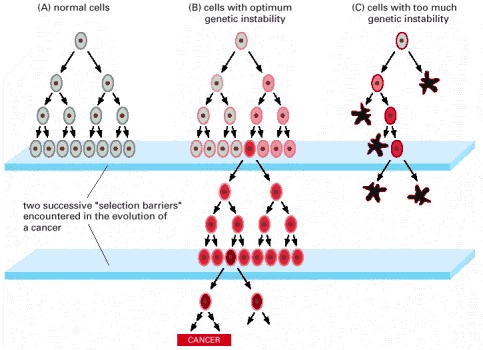
Figure above shows genetic instability and tumor progression. Cells that maintain an optimum level of genetic instability may be the most successful in the race to form a tumor. (A) In normal cells, the intrinsic amount of genetic instability is low. Thus when normal cells hit a selection barrier—low levels of oxygen or a scarcity of proliferation signals, for example—they are very unlikely to be mutable enough to produce a cell that continues proliferating. (B) In tumor cell precursors, an increased level of genetic instability makes it likely that at least one cell will contain the requisite genetic alteration to pass the selection barrier and continue the process of tumor progression. This genetic instability is retained in the lineage, and it can be measured in the resulting tumor. (C) If the level of genetic instability is too high, many of the cells suffer deleterious mutations and either proliferate much more slowly than their neighbors or are eliminated by cell death. This excessive mutability can lead to extinction of the cell lineage.
_______
From somatic mutation theory to cell work theory:
Since the discovery of oncogenes and tumor suppressor genes, modern cancer research has assumed that gene mutations alone are responsible for the initiation and progression of cancer (somatic mutation theory; Weinberg, 2014). Although this theory has been considered the central dogma of carcinogenesis for almost 40 years, no proof has yet been provided that it is causal for all malignant tumors (Soto and Sonnenschein, 2004; Soto and Sonnenschein, 2011; López-Lázaro, 2018; Supplementary Material S1). Hanselmann and Welter assume that mutations are not conditio-sine-qua-non, but that they are sufficient, but not necessary, for cancer cells to develop. In 2016, they published a hypothesis on the cause of cancer (Hanselmann and Welter, 2016), in which authors attempted to clarify two things:
(1) Mutations cannot be considered the sole cause of cancer initiation.
(2) As an equal cause of the initiation of cancer cells, changes in the energy balance (energy) or the composition of the intra- and extracellular molecules (matter/milieu) can be responsible in addition to certain mutations in cancer genes. In the publication, authors discussed that cells must essentially be regarded as complex systems that are determined by three influencing variables: energy, matter, and information.
_
If one places the cell work with the four overarching influencing variables proposed by authors (energy, matter, information, and mechanics = EMIM) at the center in order to describe and understand the processes of life, a mental model emerges that makes it possible to comprehend life mechanisms and the complex processes of initiation and progression of cancer. Authors descriptively summarize the four EMIM influencing variables of the mechanisms that condition the respective cell works.
(1) Energy: The flow of (physical and biological) energy is the driving force of all cell work and is therefore a mandatory prerequisite for the development of life processes.
(2) Matter (milieu): For the individual reactions and processes to take place properly, all the necessary molecules (proteins, RNA, electrolytes, and other reaction partners or other milieu partners, i.e., protons) must be present in the required concentration.
(3) Information: For instance, for the proteins involved to be present correctly, it is necessary for the coding DNA to be free from mutations and for protein synthesis to proceed properly.
(4) Mechanics: For the synthesis of ATP from ADP and phosphate, for example, the rotation (molecular mechanics) of parts of the enzyme complex (ATP synthetase) is necessary.
_
Authors present the following findings, which they assume, show that cell functions are not exclusively genetically determined, but are a consequence of the aforementioned molecular interactions, reactions, and biophysical influences that act on the molecules in the immediate environment and make it difficult to predict their function:
(1) In addition to its telomere-extending function, human telomerase (hTERT) can also function as a reverse transcriptase and an RNA-dependent RNA polymerase (RDRP) (Machitani et al., 2020; Yasukawa et al., 2020). The fact that an enzyme can perform such different functions is remarkable and raises the question of how these functions are regulated.
(2) Glycolysis enzymes are another example of this. There is increasing evidence that most glycolytic enzymes are deregulated in cancer cells and play an important role in tumorigenesis. Recent studies have shown that all essential glycolytic enzymes are translocated to the nucleus, where they are involved in tumor progression, regardless of their recognized metabolic function. These non-classical properties include anti-apoptotic functions, regulation of epigenetic modifications, modulation of transcription factors and cofactors, a role as extracellular cytokines, a protein kinase activity, and involvement in the mTorc1 signaling pathway (Kim and Dang, 2005; Gough, 2008; Tristan et al., 2011; Gao et al., 2012; Hamabe et al., 2014; Seki and Gaultier, 2017; Yu and Li, 2017). This makes it clear that these multifaceted glycolytic enzymes not only fulfil their classic tasks but can also perform a wide variety of functions. Could the properties be regulated only by genes or is it not possible that they are caused by a local milieu and stochastic processes?
(3) In a review article, Sherr and Roberts (2004) discuss that cyclins or cyclin-dependent kinases are not strictly essential for cell cycle progression. In this paper, he presents several knockout mouse experiments in which one or more of these proteins were deleted. The results showed that embryos have largely developed normally. Others died in utero, but not with multiple morphological changes, as expected, but in some cases with only single abnormalities.
(4) In recent years, it has become clear that transcription factors involved in transcription are intrinsically disordered proteins that have special properties (high structural flexibility, interaction with a wide variety of binding partners, etc.). These highly flexible proteins are strongly influenced by the cell milieu (Alberti et al., 2019), for example, ATP concentration, pH, or the molecular composition of the milieu, to name but a few examples (Patel et al., 2017; Garcia-Cabau and Salvatella, 2021; Mehringer et al., 2021). Because of these properties, these proteins do not act according to the classical key/lock principle but follow stochastic mechanisms (Raj and van Oudenaarden, 2008; Chubb, 2017). It follows that the simplified description of transcription by protein/protein and protein/DNA interactions is not appropriate.
_
Against this background, authors believe it will be an exciting question to determine whether and to what extent altered EMIM influencing variables contribute to shifts in protein and cell function. That this is possible in principle is already shown by generally accepted influences such as the pH value, the milieu, and the temperature on the enzyme activity or the protein structure.
Furthermore, it became apparent that cell work should be at the center of the observation and evaluation of cells. In their view, this biophysical parameter is suitable for describing cell functions and examining and explaining them in the context of biological systems, such as functional substructures, organelles, and cells. Against this background, they discuss that cellular work is not only influenced and possibly disturbed by one variable, namely genes (information) and their derivatives, but also by other factors, such as energy, matter/milieu, and mechanics. This approach makes it clear that the cause of cancer can also be initiated and driven by parameters other than genes.
_
Another aspect seems important to us at this point. If proteins can perform many fundamentally different functions, it is conceivable that if the function of one protein is disrupted, this function can be (partially) compensated for by another protein. Because of their homology, some proteins in a family can substitute for each other (Sherr and Roberts, 2004). However, if proteins can perform versatile functions, this would mean that a defined protein does not necessarily have to be at its site of action, as its function can be compensated for by other proteins (Sherr and Roberts, 2004; Yoon et al., 2012). In this context, intrinsically disordered proteins may be of particular interest (Wallmann and Kesten, 2020).
_
In a nutshell:
Cell work is a distinguishing feature of cells. It gives the cell shape and function, generates the energy it needs, and produces progeny to ensure its continuation. The continuous changes produced by cell work necessitate a constant influx of biological energy. To date, molecular biology has suggested that the processes required for this are primarily regulated by genes. This view does not do justice to the complex events that take place; cell work can be described by four overarching EMIM influencing variables. These are matter, information, mechanics, and energy. Cell work is based on the mechanisms and processes of the entity of molecular interactions, which are characterized by complex molecular interactions and biophysical influences.
To understand the mechanisms of cancer development, it is necessary to examine the defined systems. Such systems can be organisms, cells, organelles, or a single reaction pathway. To evaluate the characteristics of a system, it is useful to know the cell work that contributes to the system. This can be a single or multiple work process, depending on the system under consideration, and should be the subject of future research.
Therefore, any disturbance of cell work can be described by the EMIM quantities, making several scenarios identifiable:
(1) The change is reversible.
(2) The change is irreversible, depending on the extent, which leads to two events:
- The affected areas are permanently changed and lead to a permanent shift in the affected mechanisms, and as a result, dependent processes must adapt appropriately, which can lead to further changes. As further influences constantly act on the cell in the course of life, it is modified further and further. In particular, these changes can lead to the development of chronic diseases or cancerous cells.
- Severe changes lead to cell death.
_______
_______
Cancer is difficult to cure:
Recent advances in cancer research have revealed much more about just how complex cancer is and how difficult it is to completely cure.
Understanding the keys to cancer cell growth and survival:
Cancer cells, even within the same tumor, can be different in important ways. The technical term is heterogeneous, and the consequences of heterogeneity started coming into focus only a few years ago. At that time, researchers showed that cells collected from four different regions of the same kidney cancer tumor were in fact quite different. Further studies have reinforced this finding, and cancer cell heterogeneity is now widely recognized. Given that biopsies are typically taken from a single spot within a tumor, this fact has serious implications for improving diagnostics and therapies. It also indicates that any one targeted therapy is highly unlikely to eliminate all cancer cells by itself.
What makes cancer cells survive and thrive? Evolution.
Cancer cells grow and divide with extreme rapidity and must endure a certain amount of stress and damage to their DNA. Fast-growing cancers depend on a fine balance between DNA damage and repair, but genetic changes add up over time, and the result is like evolution at warp speed, where growth-promoting mutations lead to even more rapid expansion. This contributes to the heterogeneity discussed above. It also means the cancer you find today may differ from the one you try to treat in the weeks and months to come. With modern sequencing and analysis, it’s now possible to track cancer cell evolution and begin to predict the changes before they occur. Nonetheless, it’s much harder to hit a moving target than a stationary one, and even a highly effective, precisely targeted combination of therapies may not succeed if enough cancer cells survive initial treatment and further evolve.
Structural variation in cancer-causing genes:
Genome sequences tell us a lot, but structural variations are also key players in health and disease. Structural variants include duplications, deletions, inversions and insertions of stretches of DNA, plus epigenetic changes that don’t change the sequence per se but can have significant consequences. While most structural variants are hard to detect and details about them are just beginning to emerge, the role of a particular structural variant in cancer has been known for a very long time. Researchers discovered the famous Philadelphia chromosome, which gives rise to chronic myeloid leukemia (CML), in 1960. Structural variants add to the list of genetic changes that can tip the balance toward cancerous growth through overexpression of duplicated oncogenic (cancer-causing) genes, under expression of deleted cancer suppressor genes and other insertions/translocations giving rise to oncogenic proteins.
Cancer cells can hide in plain sight:
Cancer cells, although different in many ways from other cells in the body, are known to evade our immune system or suppress key elements of the usual immune response. In some cases aggressive cytotoxic (killer) T cells — the immune cells that locate and kill invading pathogens — actually infiltrate tumors. For some reason, however, they soon halt their attack through a combination of cell-to-cell signaling and an influx of T regulator cells, a different type of immune cells that suppress the immune response. Other research found that a chemical compound is sometimes added to cancer cell DNA and suppresses the activity of certain genes, making the cells much less likely to be targeted by the immune system. By controlling the activity of these genes, cancer is able to hide in plain sight within the body and avoid an immune response.
_____
_____
Cancer Clusters:
People may become concerned that there’s a cancer cluster in their community if they believe there is a higher-than-normal number of cancers in the area. Often there’s a concern that the cancers might be caused by some type of carcinogen (cancer-causing agent) in the environment.
Scientists have a specific definition of a cancer cluster. The US Centers for Disease Control and Prevention (CDC) and the National Cancer Institute (NCI) define a cancer cluster as a greater-than-expected number of cancer cases that occurs within a group of people in a defined geographic area over a specific period of time.
_
When considering if a cancer cluster might exist, it’s important to keep in mind that cancer is common. Well over a million new cancers are diagnosed every year in the United States alone, and nearly 4 out of 10 people in the United States will develop cancer during their lifetimes. So, it’s not uncommon for several people in a relatively small area to develop cancer around the same time.
Even if the excess number of cases reported in a cancer cluster looks significant based on statistics, it doesn’t necessarily mean that the cancers are caused by something unique to that area. Some clustering of cancer cases happens by chance, but people tend to notice and report situations where rates seem to be above average.
If the excess cases of cancer don’t seem to be random, they might need to be looked at more closely to find out if they might have a common cause. Studying cancer clusters allows scientists to identify areas of increased cancer risk, as well as to try to figure out what is causing the increase in risk. For example, studying clusters of malignant mesothelioma led to the discovery of the link between asbestos exposure and this rare cancer.
For most well-documented cancer clusters that have been found to be caused by a shared exposure, the exposure took place in the workplace, rather than in the communities where people lived. Workplace exposures may be more likely to cause cancer because the level of exposure can be higher and might last longer than in other settings. Workplace exposures can also be easier to identify because the group of exposed people is better defined and easier to trace as compared to groups in the community. This is why the links between cancer and many cancer-causing agents (carcinogens) are often first found in studies of workers. Of course, it’s also possible for cancer clusters to occur in communities as well.
_
What are the possible outcomes of a cluster investigation?
There are 3 main possible outcomes from a cancer cluster investigation:
- In most cases, an investigation will show that the suspected cluster is not a true cancer cluster.
- Less often, an investigation finds a true cancer cluster, but no cause can be found.
- Rarely, an investigation finds a cancer cluster where the cause can be determined.
To help illustrate this point, in a scientific review of over 500 cancer cluster investigations done over 20 years, only about 1 in 8 found a true increase in cancer rates, and in only one case was a clear cause for the increase found.
_
Is Cancer contagious?
Cancer is NOT contagious.
You cannot “catch” cancer from someone else. Close contact or things like sex, kissing, touching, sharing meals, or breathing the same air cannot spread cancer. Cancer cells from someone with cancer are not able to live in the body of another healthy person. The immune system finds and destroys foreign cells, including cancer cells from another person.
The fact that cancer might happen more often in certain families does not mean that the family members have spread cancer to each other. Reasons for this include:
- Family members share the same genes.
- Families may have similar unhealthy lifestyles (diet and smoking, for example).
- Family members might all be exposed to the same cancer-causing agent.
_
Cancer transfer during organ transplant:
In very rare cases, cancer cells from an organ donor have caused cancer to grow in the person who got the organ. This does not happen often, because our immune systems look for cells that are not our own and destroys them. However, people who get organ transplants must take medicines to weaken their immune systems so their body doesn’t attack and destroy the transplanted organ. Organ donors are carefully screened for cancer to reduce this risk.
Still, recent studies have shown that cancer is more common in people who get organ transplants. This is likely because of the drugs given to reduce the risk of organ rejection, rather than cancer spreading from the donated organ. Because these drugs weaken the immune system, they can prevent the body from finding and attacking damaged cells and viruses that can lead to cancer.
_
Cancer transfer during pregnancy:
Even if a woman has cancer during pregnancy, the cancer rarely affects the baby. Some cancers can spread from the mother to the placenta (the organ that connects the mother to the baby), but most cancers cannot affect the baby itself.
______
______
Second Cancer:
When a person who already had cancer develops a new cancer, it is called a second cancer or second primary cancer. It is a completely new and different type of cancer than the first one.
A second cancer is not the same as a cancer recurrence. A recurrence means that the first cancer has come back, in the same area of the body or in a different area.
A second cancer may be a late effect of your first cancer or its treatment, or it may be unrelated to your first cancer. Second cancers are becoming more common since more people are living longer after their first cancer diagnosis than ever before. About 1 in every 6 people diagnosed with cancer has had a different type of cancer in the past.
_
Second primary cancers have become an increasingly important concern in oncology during the last two decades, as they now comprise the sixth most common group of malignancies after skin, colorectal, lung, breast, and prostate cancers. What was formerly a problem primarily in paediatric cancer survivors and for the survivors of the more curable adult cancers has become a more universal problem in the present-day practice of oncology. Survivors of all cancers are living for longer periods, partly because of the more frequent use of radiation and chemotherapy. The formerly unacceptable toxicities of these modalities are more readily controlled with better supportive care. These therapies, as well as underlying predisposition, have been implicated in the etiology of second neoplasms. Other reasons for an increase in multiple cancers include fewer competing causes of mortality and consequently more naturally occurring cancers in an increasingly aging population. When reviewing data on second malignant neoplasms, consideration must be given to the definition. One can readily appreciate a “second cancer” as such when the second neoplasm differs histologically, or molecularly, from the primary neoplasm. Reports and analyses of data have emphasized the importance of ruling out metastatic disease when a new tumor arises.
_
Second cancers have also been known to result from the radiation and chemotherapy used to treat a primary cancer. Studies of paediatric cancer survivors have provided much information concerning the relationship of therapy to second neoplasms because of the young age at diagnosis and the high cure rates following successful therapy. As therapy becomes more intensive for adults as well as children and cure rates increase, we may expect a resultant increase in toxicity, including second cancers. It should be emphasized, however, that most second cancers occur naturally and randomly in cancer survivors, and so are a sign of improved survival following the first cancer diagnosis. Fear of a second primary should not outweigh the use of curative therapy for the first cancer. The most common secondary neoplasm is secondary acute myeloid leukemia, which develops primarily after treatment with alkylating agents or topoisomerase inhibitors. Survivors of childhood cancer are more than 13 times as likely to get a secondary neoplasm during the 30 years after treatment than the general population. Not all of this increase can be attributed to chemotherapy. A USA follow-up study of 650,000 cancer patients found that, overall, a relatively small proportion of second cancers is related to radiotherapy in adults, suggesting that most second cancers are due to other factors.
_____
_____
Section-3
The Hallmarks of Cancer:
Thirty years of research culminated in the year 2000 in an insightful and thorough review paper by Douglas Hanahan and Robert Weinberg that synthesized our knowledge into six essential, acquired characteristics necessary for cellular transformation. These characteristics included (1) self-sufficiency in growth signals, (2) insensitivity to antigrowth signals, (3) the ability to evade apoptosis, (4) limitless replicative potential, (5) sustained angiogenesis, and (6) the capacity to invade tissues and metastasize. The importance of this paper was less in describing a list of events and more in the synthesis of these events because the concepts proposed by Hanahan and Weinberg created a paradigm shift in our understanding of cancer. Some of the important concepts that were clarified include: No single gene is universally responsible for transformation; five or six mutations are the minimum probable number required to endow the cancer phenotype (an observation that has since been confirmed experimentally); each step is regulated by multiple interactive biochemical pathways, and thus mutations of different genes along a pathway can result in equivalent phenotypes and, conversely, mutations of the same gene can result in different cancers with distinct biology; tumors behave as tissues; and the interactions between the tumor and its microenvironment are major drivers of cancer behavior.
_
In early 2011, Hanahan and Weinberg updated the hallmarks of cancer in a new review that will likely be even more influential than the first. In this “next generation,” hallmarks of cancer were refined and reassessed, and new “enabling characteristics” (genome instability and mutation, and tumor-promoting inflammation) and “emerging” hallmarks (deregulating cellular energetics and avoiding immune destruction) were added to the paradigm. The impact of this unifying conceptualization of cancer genetics and this level of understanding are clearly evident when we consider how they have influenced the design, development, implementation, and success of new cancer therapies as seen in the figure below.
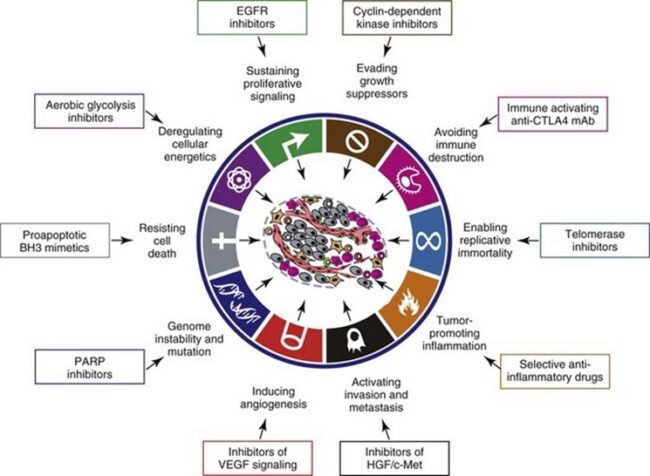
Figure above shows therapeutic targeting of the hallmarks of cancer. Drugs that interfere with each of the acquired capabilities necessary for tumor growth and progression have been developed and are in clinical trials or in some cases approved for clinical use in treating certain forms of human cancer. Additionally, investigational drugs are being developed to target each of the enabling characteristics and emerging hallmarks, which also hold promise as cancer therapeutics. The drugs listed are but illustrative examples; there is a deep pipeline of candidate drugs with different molecular targets and modes of action in development for most of these hallmarks. EGFR, Epidermal growth factor receptor; PARP, poly ADP ribose polymerase; VEGF, vascular endothelial growth factor; HGF, hepatocyte growth factor.
___
-1. Self-Sufficiency of Growth Signals:
Arguably, the most important event in neoplastic transformation is the capability of cells to sustain chronic proliferation. Under normal conditions, cells communicate with each other and integrate environmental signals by sensing cues and gradients. For example, migration, metabolism, and proliferation of mature hematopoietic cells are regulated in autocrine and paracrine fashions by locally secreted cytokines. The same cytokines may travel systemically and act in an endocrine fashion. Generally, the cytokines work by binding transmembrane receptors, which in turn initiate signaling cascades that culminate in transcriptional changes that allow the cell to adapt its behavior to match the environmental signal. The activity of these cytokines, their receptors, and the corresponding signaling molecules are finely tuned. The system can be shut down when the concentration of the cytokine falls below a threshold that can stably bind the receptor, when the receptor ceases to be expressed, or when signaling molecules are downregulated or otherwise inactivated. However, mutations in even one of the molecules involved in regulating these pathways can provide sustained growth signals in the absence of the initiating cytokine. Among many examples, there is a translocation between chromosome 2 and chromosome 5 (t(2;5)) that is present in almost half of human anaplastic lymphomas. The translocation creates a fusion protein between the nucleophosmin gene (NPM1) and the anaplastic lymphoma kinase gene (ALK), which aberrantly activates the Jak2/STAT5 signaling pathway that is normally responsive to various interleukins (IL), including IL-2, IL-3, and IL-6. The genes that encode the normal growth-promoting proteins (such as ALK, Jak2, and STAT5) are called proto-oncogenes; the mutated versions that allow cells to gain self-sufficiency from the environmental signals are called oncogenes. It is important to note that not all growth-promoting genes have the capacity to become oncogenes and that the outcomes of oncogenic activation are most commonly senescence or apoptosis, unless there are additional events that promote stable transformation and survival.
__
-2. Insensitivity to Antigrowth Signals:
In addition to the hallmark capability of inducing and sustaining positively acting growth-stimulatory signals, cancer cells must also circumvent powerful programs that negatively regulate cell proliferation; many of these programs depend on the actions of tumor suppressor genes. To maintain homeostasis, cells also must integrate antigrowth signals from the environment. Quiescence in nonhematopoietic cells is enforced by signals delivered by contact inhibition. Hematopoietic cells, on the other hand, utilize cell-cell contacts to maintain interactions within the niche and to regulate the timing and intensity of hematopoiesis, inflammation, and immunity.
“Stop” signals are usually delivered and integrated by the products of tumor suppressor genes, which derive their name largely from the observation that their inactivation facilitates tumor formation. Tumor suppressor genes balance the activity of growth-promoting proto-oncogenes and tend to act in tandem with these in most biochemical pathways. Loss of function of one or more tumor suppressor genes occurs in virtually every cancer, with inactivation of p53, RB1, PTEN, or CDKN2A each seen in more than 50% of all tumors. Each of these pathways may contribute to the pathogenesis of bone marrow–derived tumors in companion animals, and their dysfunction also may be predictive for outcomes.
__
-3. Evasion of Cell Death:
Apoptosis, or programmed cell death, is the imprinted outcome for every cell in multicellular organisms. Survival requires support from extrinsic (environmental) factors, as well as precise balance of cellular energetics and metabolism. Bone marrow–derived cells normally undergo apoptosis when concentrations of survival factors (e.g., stem cell factor, IL-3, IL-7) or nutrients are limiting or when there are severe disruptions to cellular bioenergetics.
Many cells undergo programmed cell death, or apoptosis, during fetal development. Apoptosis also may occur when a cell becomes damaged or deregulated, as is the case during tumour development and other pathological processes. Thus, when functioning properly, the body can induce apoptosis to rid itself of cancer cells.
Not all cancer cells succumb in that manner, however. Some find ways to escape apoptosis. Two mutations identified in human tumours lead to a loss of programmed cell death. One mutation inactivates the p53 gene, which normally can trigger apoptosis. The second mutation affects a proto-oncogene called BCL-2, which codes for a protein that blocks cell suicide. When mutated, the BCL-2 gene produces excessive amounts of the BCL-2 protein, which prevents the apoptosis program from being activated. Malignant lymphomas that stem from B lymphocytes exhibit this BCL-2 behaviour. The alteration of the BCL-2 gene is caused by a chromosomal translocation that keeps the gene in a permanent “on” position. Loss of p53 function protects cells from only certain kinds of suicide, whereas the BCL-2 alteration completely blocks access to apoptosis.
The blocking of apoptosis is thought to be an important mechanism in tumour generation. That mutation also may contribute to the development of tumours that are resistant to radiation and drug therapies, most of which destroy cancer cells by inducing apoptosis in them. If some cells within a tumour are unable to commit suicide, they will survive treatment and proliferate, creating a tumour refractory to therapy of this type. In this way apoptosis-inducing therapies may actually select for cancer cells resistant to apoptosis.
A more recent concept in the cell death field is autophagy—a process that tumor cells have efficiently coopted as a means to survive under adverse conditions. As part of the autophagy program, intracellular vesicles termed autophagosomes surround intracellular organelles and fuse with lysosomes. There, the organelles are broken down and then are channeled to form new molecules that support the energy-producing machinery of the cell, allowing it to survive in the stressed, nutrient-limited environment that defines most cancers.
Tumor cells also must avoid death by anoikis or loss of integral cell-to-cell or cell-to-matrix contacts. Absent these physiologic death pathways, the body often reacts to the anatomic and physiologic disruptions caused by cancer cells by targeting these cells for destruction through inflammatory pathways. This is but one pathway that leads to necrosis, since it appears that the process of necrosis also might be regulated genetically, providing another mechanism that favors survival of the whole (organism or tumor) over survival of the one. We are probably on the edge of an explosion of new findings that will further nuance our perception of how evasion (or inciting) of these cell death mechanisms contributes to neoplastic transformation and tumor progression.
__
-4. Limitless Replicative Potential:
Immortalization is another essential feature of cancer. The genetic program limits the number of times a cell is able to replicate, the so-called Hayflick limit, and when this limit is reached, replicative senescence is induced. Induction of replicative senescence does not induce death; cells maintain energetic homeostasis and remain functional, but they undergo significant genetic changes characterized by telomere erosion. Cells that are able to replicate must maintain the integrity of telomeres, which are “caps” made of repetitive DNA sequence that protect chromosomes from destruction. Solid tumors acquire immortalization predominantly by activation of the telomerase enzyme system and the consequent maintenance of telomere integrity. In hematopoietic cells, telomerase activity seems to be retained longer than in other somatic cells, so it is possible this facilitates immortalization in lymphoma and leukemia. The role of immortalization and the importance of telomerase (both to maintain telomere length and to maintain other biochemical functions that are essential for cell survival) are well established; however, the role of replicative senescence has recently been questioned because improved technology has allowed researchers to circumvent this process in normal cells.
____
-5. Sustained Angiogenesis:
Angiogenesis plays a critical role in the growth of cancer because solid tumors need a blood supply if they are to grow beyond a few millimeters in size. Tumors can actually cause this blood supply to form by giving off chemical signals that stimulate angiogenesis. Tumors can also stimulate nearby normal cells to produce angiogenesis signaling molecules. The resulting new blood vessels “feed” growing tumors with oxygen and nutrients, allowing the tumor to enlarge and the cancer cells to invade nearby tissue, to move throughout the body, and to form new colonies of cancer cells, called metastases.
_
Angiogenesis is a highly regulated process, whereby new blood vessels form from preexisting ones. In adult mammals, physiologic angiogenesis is largely limited to the ovaries, uterus, and placenta, with the turnover rate of vascular endothelial cells being very low in most other tissues. Pathophysiologic angiogenesis is a characteristic of wound healing and diseased states, particularly cancer, where the number of proliferating endothelial cells increases significantly and the morphology of the vasculature is altered in multiple ways. For many types of cancer, as tumor cells undergo dysregulated proliferation, the tumor mass initially expands beyond the support capacity of the existing vasculature, leading to decreased levels of oxygen and nutrients and the accumulation of metabolic wastes. Tumor cells respond to this deterioration of the tumor microenvironment by up-regulating several proangiogenic factors, including vascular endothelial growth factor (VEGF)-A, basic fibroblast growth factor, placental growth factor, and platelet-derived endothelial growth factor, which collectively activate quiescent endothelial cells and promote their migration into the tumor. This shift of the tumor microenvironment to an angiogenic state, or ‘‘angiogenic switch’’, is an important rate limiting factor in tumor development. Despite the active angiogenesis induced by tumor cell-derived proangiogenic factors, structural defects associated with the tumor vasculature often lead to inefficient blood perfusion in established tumors, which contributes to tumor hypoxia. Tumor metastasis is also regulated by angiogenesis, as well as by lymphangiogenesis, where new lymphatic vessels are formed from preexisting ones. Tumor cell dissemination, the first step in tumor metastasis, requires access to both blood and lymphatic circulation. Once successfully extravasated, the survival and further colonization of the disseminated tumor cells is dependent on angiogenesis at the secondary site. Angiogenesis is thus a key factor in the development and metastasis of a variety of tumor types and is an important hallmark of malignant disease. Moreover, angiogenesis presents unique opportunities for therapeutic intervention in cancer treatment, as first proposed by the late Judah Folkman more than 35 years ago.
_
The process of angiogenesis requires the coordinated action of a variety of growth factors and cell-adhesion molecules in endothelial and stromal cells. So far, vascular endothelial growth factor-A (VEGF) and its receptors comprise the best-characterized signaling pathway in tumor angiogenesis. VEGF binds several receptor tyrosine kinases, including VEGF receptor-1 (VEGFR-1 [Flt-1]) and VEGFR-2 (KDR, Flk-1). VEGFR-2 is the major mediator of the angiogenic effects of VEGF. However, VEGFR-1 is expressed by some tumor cells and may mediate chemotactic signals, thus potentially having a role in cancer growth. The expression of VEGF is upregulated by hypoxia and inflammation. The transcription factor hypoxia-inducible factor-1α (HIF), which is part of a pathway that also includes regulation by the von Hippel-Lindau (VHL) tumor suppressor gene, is a major regulator of VEGF expression. Under conditions of normal oxygen tension, VHL protein targets HIF for degradation; under low oxygen conditions, HIF increases as VHL-mediated degradation is reduced, allowing for upregulation of VEGF. Other signaling molecules also contribute to angiogenesis, including platelet-derived growth factor-β (PDGF-β) and its receptor (PDGFR), and the angiopoietins. PDGF-β is required for recruitment of pericytes and maturation of new capillaries. Recent studies also document the importance of tumor-derived PDGF in recruitment of stroma that produces VEGF and other angiogenic factors as seen in figure below.
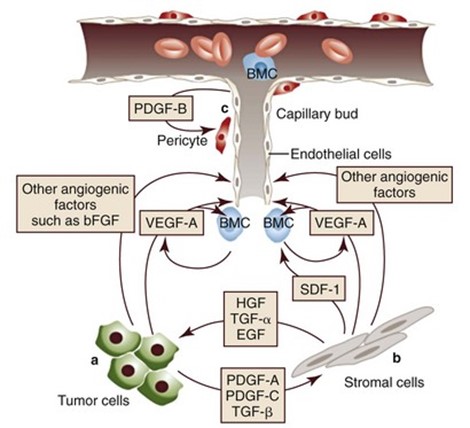
Figure above shows few of the molecular and cellular players in the tumor/microvascular microenvironment. A, Tumor cells produce VEGF-A and other angiogenic factors, such as bFGF, angiopoietins, interleukin-8 (IL-8), placenta growth factor (PlGF), and VEGF-C. These stimulate resident endothelial cells to proliferate and migrate. B, An additional source of angiogenic factors is the stroma, which is a heterogeneous compartment, comprising fibroblastic, inflammatory, and immune cells. Recent studies indicate that tumor-associated fibroblasts produce chemokines such as SDF-1, which may recruit bone marrow–derived angiogenic cells (BMC). VEGF-A or PlGF may also recruit BMC. Tumor cells may also release stromal cell-recruitment factors, such as PDGF-A, PDGF-C, or transforming growth factor (TGF-α, TGF-β). A well-established function of tumor-associated fibroblasts is the production of growth/survival factor for tumor cells such as EGFR ligands, HGF, and heregulin. C, Endothelial cells produce PDGF-β, which promotes recruitment of pericytes in the microvasculature after activation of PDGF receptor-β (PDGFR-β) factor. VEGF-A, Vascular endothelial growth factor-A; bFGF, basic fibroblast growth factor; SDF-1, stromal cell–derived factor-1; HGF, hepatocyte growth factor; PDGF, platelet-derived growth factor; EGFR, epidermal growth factor receptor.
_
The control of tumor angiogenesis is an integral part of the host defense response to tumor growth. Loss of endogenous angiogenesis inhibitors, such as endostatin and thromobospondin-1, leads to increased tumor angiogenesis and accelerates tumor growth in transgenic mouse models. In contrast, when angiogenesis is impaired and the expression of endogenous angiogenesis inhibitors is increased, tumors may enter a period of prolonged dormancy. Dormant tumors may be found in autopsy samples from trauma victims and in cancer patients that relapse after being disease-free for months or even years, indicating that tumors can exist as microscopic lesions for long periods without any clinical manifestation of disease. This dormancy may reflect the inability of these in situ tumors to disrupt or circumvent host antiangiogenic defenses. The inhibition of tumor growth by antiangiogenic drugs has been achieved in both preclinical studies and clinical trials, where promising antitumor responses have been reported for a variety of antiangiogenic agents.
___
-6. Invasion and Metastasis:
The role of genetic events in invasion and metastasis is still incompletely understood. The classic model of metastasis proposed by Fidler suggests a step-wise acquisition of assets that enables cells to leave the primary tumor site, travel through blood or lymph, invade stroma in favorable locations, and thus become reestablished at distant sites. More recent work suggests that most tumors possess the ability to dislodge cells that travel to distant sites, and the ability of such cells to survive in capillary beds may be the most important step in the metastatic process.
Bone marrow–derived cells have intrinsic properties that allow them to travel throughout the body, traffic through all major organs, and home to areas of inflammation. Thus bone marrow–derived tumors are inherently metastatic. Nevertheless, hematopoietic tumors that are cytologically indistinguishable can have distinct and preferential tissue distribution. We do not fully understand what events make leukemic cells stay in the peripheral circulation, while cells from corresponding lymphomas (or myeloid sarcomas) with virtually identical molecular signatures stay confined to lymphoid or visceral organs.
In epithelial neoplasms that account for the majority of tumors in humans, the epithelial-to-mesenchymal transition (EMT) has received increasing attention for its role in metastasis. EMT is a developmental program that progenitor stem cells use during morphogenesis and can be physiologically reactivated during wound healing. It remains unclear whether EMT is equally (or less) important in sarcomas more commonly seen in domestic animals, in which the cells of origin seem to retain EMT capabilities to a greater extent. Similarly, there is increasing evidence of interactions between cancer cells, the “initiating” population in the tumor (cancer stem cells), mesenchymal stem cells (MSCs), tumor-associated fibroblasts, inflammatory cells, and angiogenic cells, which may be responsible for invasive behaviors and possibly for survival in hostile environments that exist at distant sites as seen in figure below.
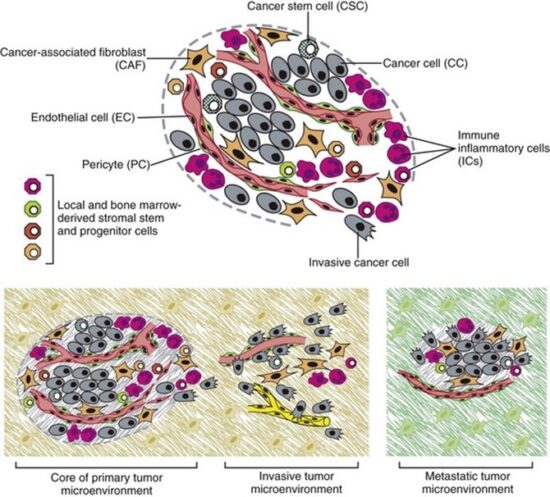
Figure above shows cells of the tumor microenvironment.
Upper, An assemblage of distinct cell types constitutes most solid tumors. Both the parenchyma and stroma of tumors contain distinct cell types and subtypes that collectively enable tumor growth and progression. Notably, the immune inflammatory cells present in tumors can include both tumor-promoting, as well as tumor-killing, subclasses.
Lower, The distinctive microenvironments of tumors. The multiple stromal cell types create a succession of tumor microenvironments that change as tumors invade normal tissue and thereafter seed and colonize distant tissues. The abundance, histologic organization, and phenotypic characteristics of the stromal cell types, as well as of the extracellular matrix (hatched background), evolve during progression, thereby enabling primary, invasive, and then metastatic growth. The surrounding normal cells of the primary and metastatic sites, shown only schematically, likely also affect the character of the various neoplastic microenvironments.
(Not shown are the premalignant stages in tumorigenesis, which also have distinctive microenvironments that are created by the abundance and characteristics of the assembled cells.)
_
Adaptive Evolution and the Tumor Microenvironment:
There is a bidirectional flow of information between the tumor and the microenvironment, with each helping to mold the other into functional growing tissue that can evade or withstand attack by the host. The reference to a “selective growth advantage” that is reminiscent of Darwinian selection is not accidental. The clonal evolution theory addresses the significance of sequential genetic changes providing growth and survival advantages, but to this we must add the fact that, in addition to these self-sufficient events that influence growth and survival, tumor cells must also evade “predators” (e.g., inflammation and the immune system). In essence, the interaction of the tumor with its microenvironment and ultimately with the host is in fact subject to Darwinian laws of evolution, albeit in an accelerated time scale. This is evident in the ability of tumors to modulate stromal cells to support their own growth by providing a suitable matrix and an abundance of nutrients, while maintaining antitumor responses at bay.
______
______
Emerging and Enabling Hallmarks:
-1. Genomic Instability:
The concept of genomic instability is not new but was incorporated as an “enabling hallmark” into the updated Hanahan and Weinberg model. Step-wise clonal evolution is satisfying because it can be correlated with discrete pathologic changes in tumor progression, especially for epithelial tumors where such progression can be seen in lesions that go through stages of hyperplasia, atypical hyperplasia (dysplasia), adenoma, carcinoma in situ, invasive carcinoma, and metastatic carcinoma. However, analysis of tumor genomes even in early stages usually shows aneuploidy (abnormal DNA copy number), as well as chaotic changes indicative of multiple numeric and structural DNA abnormalities. Similar abnormalities first noticed by Boveri more than 100 years ago in studies of sea urchin cells led him to formulate the “aneuploidy theory” of cancer. We know now that aneuploidy is especially evident in solid tumors; based on this, Loeb proposed the existence of the “mutator phenotype” in which cells are predisposed to undergo multiple mutations, some of which inevitably lead to cancer. Some tenets of his hypothesis appear to be correct, although perhaps in different circumstances than Loeb originally envisioned, because they might relate to increased activity of polymerases with low fidelity under conditions in which the rate of DNA damage (and consequently mutations) is higher than the expected background from normal DNA replication (e.g., in lung epithelial cells from heavy smokers). However, direct measurements of mutation rates of sporadic tumors are much lower than those predicted if a “mutator phenotype” was operative in these tumors. Indeed, the minimum number of mutations that are required for clinical onset of cancer in solid tumors based on sequencing of solid tumor genomes is 15 to 25, but this may apply to tumors with chaotic karyotypes, as the number of mutations identified in a cytogenetically stable leukemia was significantly smaller.
Still, genetic instability is a hallmark of most tumors, and while it can be partly explained by increased errors in DNA replication and chromosomal segregation in cells that are rapidly dividing, other mechanisms are clearly operative, involving telomeres and telomerase. Although many of these changes are not “recurrent” and appear to be random products of instability, some may in fact contribute to proliferative crisis. It is possible that the initiation events for many tumors occur early in life during highly proliferative stages of tissue growth and remodeling (e.g., prior to closure of the growth plates in bone cancer), but they become evident later in life when a last series of mutations allows the transformed cell to reach this crisis stage. Hematopoietic tumors seem to avoid the chaotic chromosomal instability associated with solid tumors. We do not fully understand the reasons for this, although it may be partly due to intrinsic protective mechanisms associated with the proliferative rate of bone marrow precursor cells.
___
-2. Tumor-Promoting Inflammation:
The role of inflammation in cancer has received considerable attention in the past 10 years. Although our understanding of this phenomenon remains incomplete, it clearly met the criteria for inclusion as an “enabling hallmark” into the updated Hanahan and Weinberg model. The importance of inflammation was inferred from the earliest microscopic studies of cancer, but it was a seminal paper by Dvorak in 1986, in which he described tumors as “wounds that never heal,” that provided synthesis for the recurrent observation that tumors were often infiltrated by inflammatory cells of the innate (granulocytes, histiocytes, and macrophages) and the adaptive (lymphocytes) immune systems. Mechanistic distinctions remain to be defined between inflammation that favors tumor growth and inflammation that retards growth or eliminates the tumor, but we can say confidently that inflammation contributes to tumor growth and survival by supplying factors that sustain proliferation; factors that limit cell death; proangiogenic factors; extracellular matrix-modifying enzymes that facilitate angiogenesis, invasion, and metastasis; and other signals that lead to activation of EMT and other hallmark-facilitating programs. Inflammatory cells also release notably reactive oxygen species that are actively mutagenic for nearby cancer cells, accelerating their genetic evolution toward states of heightened malignancy.
_
Inflammatory conditions in selected organs increase the risk of cancer. An inflammatory component is present also in the microenvironment of tumors that are not epidemiologically related to inflammation. Recent studies have begun to unravel molecular pathways linking inflammation and cancer. In the tumor microenvironment, smouldering inflammation contributes to proliferation and survival of malignant cells, angiogenesis, metastasis, subversion of adaptive immunity, reduced response to hormones and chemotherapeutic agents. Recent data suggest that an additional mechanism involved in cancer-related inflammation (CRI) is induction of genetic instability by inflammatory mediators, leading to accumulation of random genetic alterations in cancer cells.
_
Epidemiological studies have revealed that chronic inflammation predisposes to different forms of cancer. Usage of non-steroidal anti-inflammatory agents is associated with protection against various tumors, a finding that to a large extent mirrors that of inflammation as a risk factor for certain cancers. The ‘inflammation–cancer’ connection is not restricted to increased risk for a subset of tumors. An inflammatory component is present in the microenvironment of most neoplastic tissues, including those not causally related to an obvious inflammatory process. Key features of cancer-related inflammation (CRI) include the infiltration of white blood cells, prominently tumor-associated macrophages (TAMs); the presence of polypeptide messengers of inflammation [cytokines such as tumor necrosis factor (TNF), interleukin (IL)-1, IL-6, chemokines such as CCL2 and CXCL8] and the occurrence of tissue remodeling and angiogenesis.
_
Recent efforts have shed new light on molecular and cellular circuits linking inflammation and cancer. Two pathways have been schematically identified; in the intrinsic pathway, genetic events causing neoplasia initiate the expression of inflammation-related programs that guide the construction of an inflammatory microenvironment [e.g. RET oncogene in papillary carcinoma of the thyroid]. Oncogenes representative of different molecular classes and mode of action (tyrosine kinases, ras–raf, nuclear oncogenes and tumor suppressors) share the capacity to orchestrate proinflammatory circuits (e.g. angiogenetic switch; recruitment of myelo-monocytic cells). In the extrinsic pathway, inflammatory conditions facilitate cancer development. The triggers of chronic inflammation that increase cancer risk or progression include infections (e.g. Helicobacter pylori for gastric cancer and mucosal lymphoma; papilloma virus and hepatitis viruses for cervical and liver carcinoma, respectively), autoimmune diseases (e.g. inflammatory bowel disease for colon cancer) and inflammatory conditions of uncertain origin (e.g. prostatitis for prostate cancer). Key orchestrators at the intersection of the intrinsic and extrinsic pathway include transcription factors and primary proinflammatory cytokines. Thus, CRI is a key component of tumors and may represent the additional hallmark of cancer.
_
In the extrinsic pathway, it remains uncertain whether chronic inflammation per sé is sufficient for carcinogenesis. Reactive oxygen and nitrogen intermediates are obvious inflammation-generated candidate mediators for DNA damage and evidence obtained in vitro and in vivo is consistent with this view. There are data suggesting that inflammatory cells and mediators can destabilize the cancer cell genome by a variety of mechanisms either directly inducing DNA damage or affecting DNA repair systems and altering cell cycle checkpoints. These emerging data suggest that an additional mechanism by which inflammation can contribute to cancer initiation and progression is genetic destabilization of cancer cells. The high degree of genetic heterogeneity in tumors suggests that genetic instability is an ongoing process throughout tumor development. Accumulating evidence supports the view that inflammatory mediators, some of that are direct mutagens, also directly or indirectly downregulate DNA repair pathways and cell cycle checkpoints, thus destabilizing cancer cell genome and contributing to the accumulation of random genetic alterations. These in turn accelerate the somatic evolution of cancer to a genomically heterogenous population of expanding cells naturally selected for their ability to proliferate, invade and evade host defense.
_
Finally, inflammatory reactions can also result in antitumor activity (the bright side of the force). This dual function of inflammatory cells and mediators is reflected by studies on correlations between parameters of CRI and clinical behavior in different contexts. The challenge now is to identify the mechanisms triggering a ‘bright’ inflammatory response leading to tumor inhibition, whereas neutralizing the protumor actions of the dark side.
___
-3. Reprogramming Energy Metabolism:
In the early years of the twentieth century, Otto Warburg observed that cancer cells preferentially utilized glycolytic rather than oxidative pathways to generate energy even under conditions of normal or high oxygen. This metabolic peculiarity of cancer cells, called the Warburg effect, seems to be driven by activated oncogenes and/or by loss of tumor suppressor genes that provide cancer cells with selective growth and survival advantages by conferring the hallmark capabilities of cell proliferation, avoidance of cytostatic controls, and attenuation of apoptosis. The reliance of cancer cells on glycolysis can be further accentuated under the hypoxic conditions. In fact, Warburg-like metabolism seems to be present in rapidly dividing embryonic tissues, suggesting a role in supporting large-scale biosynthetic programs that are required for active cell proliferation.
Cancer cells do not seem to enable the Warburg effect universally. Rather, much like other cells with high energetic demands, they seem to sort out into lactate-secreting (Warburg) and lactate-consuming cells, providing an efficient, albeit homeostatically disturbed, energy environment. Furthermore, it seems that oxygenation is not static in tumors but instead fluctuates temporally and regionally due to the instability and chaotic organization of tumor-associated neovasculature. Altered energy metabolism is proving to be as widespread in cancer cells as many of the other cancer-associated traits that have been accepted as hallmarks of cancer. This realization raises the question of whether deregulating cellular energy metabolism is therefore a core hallmark capability of cancer cells that is as fundamental as the six well-established core hallmarks. In fact, the redirection of energy metabolism is largely orchestrated by proteins that are involved in one way or another in programming the core hallmarks of cancer. When viewed in this way, aerobic glycolysis is simply another phenotype that is programmed by proliferation-inducing oncogenes and the designation of reprogrammed energy metabolism as an emerging hallmark seems most appropriate.
It is worth noting that this characteristic of tumor cells provides at least one important diagnostic advantage. Upregulation of the major glucose transporter, GLUT-1, is seen in virtually all tumors, making the cells efficient glucose scavengers. This can be exploited to image tumor cells noninvasively with precision by visualizing glucose uptake using positron-emission tomography (PET) with a radiolabeled analog of glucose (18F-fluorodeoxyglucose [18F-FDG]) as a reporter. The combination of PET with computed tomography (PET-CT) is now one of the most robust means to evaluate composition of tumors, minimal residual disease, and tumor-specific objective responses in patients receiving conventional and experimental therapies.
___
-4. Evading Immune Destruction:
Burnet and Thomas proposed the concept that the immune system can recognize and destroy incipient tumors (cancer immunosurveillance) in the 1950s. However, the hypothesis was far ahead of its time, and technologic obstacles impeded proof, so the theory fell into disfavor. In recent years, the immunosurveillance theory has gained traction anew because data strongly suggest that the immune system helps to maintain tumors at bay, and thus tumors must evade the immune response to survive. In its recent incarnation, the theory has been refined to incorporate the concept of immunoediting, in which the immune system destroys strongly antigenic tumor cells, providing weakly antigenic cells a survival advantage. Experimental evidence for this concept includes differences between tumors grown in immunocompetent (only weakly antigenic tumors survive) and immunocompromised mice (no selection against strongly antigenic tumors is observed), but it is unknown if immunoediting is operative in spontaneous cancers.
That the tumor microenvironment forms and maintains an immunosuppressive barrier provides more compelling evidence for the role of the immune system to limit tumor growth and metastasis. This immunosuppressive barrier includes cellular factors such as regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), and MSCs. Tregs, MDSCs, and MSCs can attack distinct and complementary antigen-specific (Tregs) and nonspecific (MDSCs and MSCs) facets of immune effector cell activation and function. Soluble factors, including transforming growth factor-β (TGF-β) and immunoglobulins, also contribute to the immunosuppressive barrier directly by disabling immune effector cells and indirectly by “educating” tumor-associated stromal cells, which in turn promotes secretion of stromal-derived factors that recruit additional inflammatory cells (tumor macrophages) and endothelial cells that further accelerate tumor growth.
______
______
Section-4
Etiology of cancer:
Cancer is caused by various genetic and environmental factors. This list includes a wide range of factors, like a gain of function of many protooncogenes or loss of function of many tumor-suppressing genes, various chemical agents like alkylating agents, and physical agents like radiations. Since the early 1900s, high rates of cancer have also been linked to occupational choices. Evidence of a variety of viruses causing cancer in humans has also emerged, for example, human T-cell lymphotropic virus, HIV, HBV, HCV, HPV, Epstein-Barr virus, etc., have appeared as the top-notch causes of various cancers. Many different lifestyle factors contribute to increasing cancer risk. Together, diet and obesity are related to approximately 30–35% of cancer deaths. Dietary recommendations for cancer prevention typically include an emphasis on vegetables, fruit, whole grains, and fish, and avoidance of processed meat, red meat, animal fats, and refined carbohydrates.
_____
Division of Cancer Epidemiology and Genetics (DCEG) investigators utilize a variety of research approaches in seeking to understand the causes of cancer.
-1. Absolute Risk Modeling
DCEG investigators in the Biostatistics Branch have developed models (such as the Gail model) for projecting the individualized absolute risk of certain types of cancer. These models have been used to counsel individual patients on their disease risk; to make more formal management recommendations, such as whether or not to take tamoxifen to prevent breast cancer; to design cancer prevention trials; and to assess the potential reductions in population absolute risk from preventive activities.
-2. Descriptive Epidemiology
The Division maintains a broad-ranging, multi-faceted program of descriptive epidemiological studies utilizing a variety of methodological approaches to identify novel risk factors, evaluate tumor heterogeneity, describe current and future trends of common and rare malignancies, and project risk for second primary cancers.
-3. Exposure Assessment Studies and Methods
Evaluating exposure-response relationships is a crucial component in determining cancer causation. For that reason, quantitative exposure assessment plays an essential role in high-quality epidemiologic investigations. DCEG investigators have developed cutting-edge tools and techniques to evaluate the reliability and validity of exposure measurements used in cohort and case-control studies of occupational, lifestyle, and environmental exposures. In addition, our investigators continually work to improve upon these well-established methods.
-4. Genomic Studies
DCEG investigates the biological basis of inherited and acquired genetic variants associated with cancer susceptibility, utilizing genome-wide association studies, exome sequencing, and candidate gene studies. DCEG investigators and their collaborators employ an array of advanced statistical methods to support these studies.
-5. Metabolomics
Metabolomics is the study of small-molecule metabolites in cells, tissues, and organisms that are present in biofluids such as plasma and urine. An emerging field of study, metabolomics has the potential to improve exposure measures and delineate mechanistic links between exposures and cancer.
-6. The Human Microbiome and Cancer
The human microbiota is the collection of all the microorganisms and bacteria that live in or on the human body, such as those present in the digestive system. In the emerging field of microbiomics, researchers study the extent and patterns of these microbes at various body sites and their influence on human health and disease. DCEG investigators are at the forefront of both methodologic and cancer association studies of the microbiome in human populations.
-7. Molecular Epidemiology
DCEG investigators use molecular epidemiology to examine the relationship of genetic and environmental risk factors to cancer etiology. Using laboratory techniques, investigators look for biomarkers of disease and use them to understand the underlying mechanisms of disease in populations.
-8. Tumor Profiling in Relation to Cancer Etiology
The recognition of cancer as a heterogeneous disease, coupled with technological advances in molecular/genomic profiling of tumors, provides epidemiologists with the opportunity to integrate tissue profiling in etiologic studies to study the carcinogenesis process, and pinpoint specific factors associated with risk for developing specific molecular or genomic cancer subtypes. DCEG investigators are also undertaking foundational research to identify novel molecular and genomic signatures in tumors linked to germline genetic variants and certain environmental exposures.
_____
After sequencing his own genome, pioneer genomic researcher Craig Venter remarked at a leadership for the twenty-first century conference, “Human biology is actually far more complicated than we imagine. Everybody talks about the genes that they received from their mother and father, for this trait or the other. But in reality, those genes have very little impact on life outcomes. Our biology is way too complicated for that and deals with hundreds of thousands of independent factors. Genes are absolutely not our fate. They can give us useful information about the increased risk of a disease, but in most cases they will not determine the actual cause of the disease, or the actual incidence of somebody getting it. Most biology will come from the complex interaction of all the proteins and cells working with environmental factors, not driven directly by the genetic code”
This statement is very important because looking to the human genome for solutions to most chronic illnesses, including the diagnosis, prevention, and treatment of cancer, is overemphasized in today’s world. Observational studies, however, have indicated that as we migrate from one country to another, our chances of being diagnosed with most chronic illnesses are determined not by the country we come from but by the country we migrate to. In addition, studies with identical twins have suggested that genes are not the source of most chronic illnesses. For instance, the concordance between identical twins for breast cancer was found to be only 20%. Instead of our genes, our lifestyle and environment account for 90–95% of our most chronic illnesses.
_
Cancer is caused by both internal factors (such as inherited mutations, hormones, and immune conditions) and environmental/acquired factors (such as tobacco, diet, radiation, and infectious organisms). The link between diet and cancer is revealed by the large variation in rates of specific cancers in various countries and by the observed changes in the incidence of cancer in migrating. For example, Asians have been shown to have a 25 times lower incidence of prostate cancer and a ten times lower incidence of breast cancer than do residents of Western countries, and the rates for these cancers increase substantially after Asians migrate to the West. The importance of lifestyle factors in the development of cancer was also shown in studies of monozygotic twins. Only 5–10% of all cancers are due to an inherited gene defect. Although all cancers are a result of multiple mutations, these mutations are due to interaction with the environment.
_
These observations indicate that most cancers are not of hereditary origin and that lifestyle factors, such as dietary habits, smoking, alcohol consumption, and infections, have a profound influence on their development. Although the hereditary factors cannot be modified, the lifestyle and environmental factors are potentially modifiable. The lesser hereditary influence of cancer and the modifiable nature of the environmental factors point to the preventability of cancer. The important lifestyle factors that affect the incidence and mortality of cancer include tobacco, alcohol, diet, obesity, infectious agents, environmental pollutants, and radiation.
_
The majority of cancers, some 90–95% of cases, are due to genetic mutations from environmental and lifestyle factors. The remaining 5–10% are due to inherited genetics. Environmental refers to any cause that is not inherited, such as lifestyle, economic, and behavioral factors and not merely pollution. Common environmental factors that contribute to cancer death include tobacco use (25–30%), diet and obesity (30–35%), infections (15–20%), radiation (both ionizing and non-ionizing, up to 10%), lack of physical activity, and pollution. Psychological stress does not appear to be a risk factor for the onset of cancer, though it may worsen outcomes in those who already have cancer. It is not generally possible to prove what caused a particular cancer because the various causes do not have specific fingerprints. For example, if a person who uses tobacco heavily develops lung cancer, then it was probably caused by the tobacco use, but since everyone has a small chance of developing lung cancer as a result of air pollution or radiation, the cancer may have developed for one of those reasons. Excepting the rare transmissions that occur with pregnancies and occasional organ donors, cancer is generally not a transmissible disease, however factors that may have contributed to the development of cancer can be transmissible; such as oncoviruses like hepatitis B, Epstein-Barr virus and HIV.
_____
_____
Common Causes:
According to the American Cancer Society and the National Cancer Institute, the most common causes and risk factors of cancer are:
- Smoking and tobacco use
- Alcohol
- Lack of physical activity
- Being overweight or obese
- Poor diet
- Sun exposure
- Radiation exposure
- Virus infections and other infections
- Exposure to cancer-causing substances
- Family history and genetics
- Chronic inflammation
- Hormones
- Immunosuppression
- Age
_____
Cancers can be caused by a number of different factors and, as with many other illnesses, most cancers are the result of exposure to a number of different causal factors. It is important to remember that, while some factors cannot be modified, around one third of cancer cases can be prevented by reducing behavioural and dietary risks.
Modifiable risk factors include:
It is important to note that income and education levels, national policies, industry tactics with vested commercial interests, and genetics, are all factors that can make it very difficult to act on a modifiable factor and change behaviour at the individual level.
Alcohol – The evidence that all types of alcoholic drinks are a cause of a number of cancers is now stronger than ever before. Alcohol can increase the risk of six types of cancers, including bowel (colorectal), breast, mouth, pharynx and larynx (mouth and throat), oesophageal, liver and stomach. The evidence suggests that in general, the most alcohol drinks people consume the higher the risk of many cancers, and that even moderate alcohol intake increases the risk of cancer.
Being overweight or obese – Excess weight has been linked to an increased risk of developing 12 different cancers, including bowl and pancreatic cancers. In general, greater weight gain, particularly as adults, is associated with greater cancer risks. In general, greater weight gain, particularly as adults, is associated with greater cancer risks.
Diet and nutrition – Experts suggest that diets and nutritional intake, particularly diets high in red meats, processed meats, salted foods and low in fruits and vegetables have an impact on cancer risks, particularly colorectum, nasopharynx and stomach.
Physical activity – Regular physical activity not only helps to reduce excess body fat and the cancer risks associated with this, but being physically active can help to reduce the risks of developing colon, breast and endometrial cancers.
Tobacco – Tobacco smoke contains at least 80 different cancer-causing substances (carcinogenic agents). When smoke is inhaled the chemicals enter the lungs, pass into the blood stream and are transported throughout the body. This is why smoking or chewing tobacco not only causes lung and mouth cancers but is also related to many other cancers. The more a person smokes, the younger they start, and the longer they keep smoking, all further increase the risk of cancer. Currently tobacco use is responsible for around 22% of cancer deaths.
Ionising radiation – Manmade sources of radiation can cause cancer and are a risk for workers. These include radon, x-rays, gamma rays and other forms of high-energy radiation. Prolonged and unprotected exposure to ultraviolet radiations from the sun, sunlamps and tanning beds can also lead to melanoma and skin malignancies. Fair skinned people, individuals with a lot of moles or who have a family history of melanoma or non-melanoma skin cancer, are at highest risk. However, people of all skin tones can develop skin cancer, including individuals with darker skin.
Work place hazards – Some people risk being exposed to a cancer-causing substance because of the work that they do. For example, workers in the chemical dye industry have been found to have a higher incidence than normal of bladder cancer. Asbestos is a well-known workplace cause of cancer – particularly a cancer called mesothelioma, which most commonly affects the covering of the lungs.
Infection – Infectious agents are responsible for around 2.2 million cancer deaths annually. This does not mean that these cancers can be caught like an infection; rather the virus can cause changes in cells that make them more likely to become cancerous. Around 70% of cervical cancers are caused by Human papillomavirus (HPV) infections, while liver cancer and Non-Hodgkin Lymphoma can be caused by the Hepatitis B and C virus, and lymphomas are linked to the Epstein-Barr virus. Bacterial infections have not been thought of as cancer-causing agents in the past, but more recent studies have shown that people who have helicobacter pylori infection of their stomach develop inflammation of the stomach lining, which increases the risk of stomach cancer.
_
Non-modifiable risk factors include:
Age – Many types of cancer become more prevalent with age. The longer people live, the more exposure there is to carcinogens and the more time there is for genetic changes or mutations to occur within their cells. In general, the risk of developing cancer appears to increase until the age of 70 to 80 and then diminish, according to the National Cancer Institute (NCI). A 2020 review suggests this may be the result of:
- less effective cell repair mechanisms that come with aging
- build-up of risk factors over the course of life
- duration of exposures to carcinogens
Genetics – Some people are unfortunately born with a genetically inherited high risk for a specific cancer (‘genetic predisposition). This does not mean developing cancer is guaranteed, but a genetic predisposition makes the disease more likely. For example, women that carry the BRCA 1 and BRCA 2 breast cancer genes have a higher predisposition to developing this form of cancer than women with a normal breast cancer risk. However, less than 5% of all breast cancer is known to be due to genes. So, although women with one of these genes are individually more likely to develop breast cancer, most cases are not caused by a high risk inherited gene fault. This is true of other common cancers where some people have a genetic predisposition – for example, colon (large bowel) cancer.
The immune system – People who have weakened immune systems are more at risk of developing some types of cancer. This includes people who have had organ transplants and take drugs to suppress their immune systems to stop organ rejection, plus people who have HIV or AIDS, or other medical conditions which reduce their immunity to disease.
______
Substantial contribution of extrinsic risk factors to cancer development, a 2016 study:
Cancers were once thought to originate from mature tissue cells that underwent de-differentiation in response to cancer progression. Today, cancers are proposed to originate from the malignant transformation of normal tissue progenitor and stem cells, although this is not uniformly accepted. Nevertheless, recent research has highlighted a strong correlation of 0.81 between tissue-specific cancer risk and the lifetime population size and cumulative number of cell divisions of tissue-specific stem cells. However, there has been extensive controversy regarding the conclusion that this correlation implies a very high unavoidable risk for many cancers that are due solely to the intrinsic baseline population size of tissue-specific stem cells. Much discussion has been made to argue against the ‘bad luck’ hypothesis, yet none offered specific alternatives to quantitatively evaluate the contribution of extrinsic risk factors in cancer development. Applying several distinct modeling approaches, authors here provide strong evidence that unavoidable intrinsic risk factors contribute only modestly (<10~30%) to the development of many common cancers.
First, authors demonstrate that the correlation between stem-cell division and cancer risk does not distinguish between the effects of intrinsic and extrinsic factors. Next, they show that intrinsic risk is better estimated by the lower bound risk controlling for total stem cell divisions. Finally, they show that the rates of endogenous mutation accumulation by intrinsic processes are not sufficient to account for the observed cancer risks. Collectively, authors conclude that cancer risk is heavily influenced by extrinsic factors. These results carry immense consequences for strategizing cancer prevention, research, and public health.
The errors occurring during the division of cells, being routes of malignant transformation, can be influenced by both intrinsic processes as well as extrinsic factors (Figure below). “Intrinsic processes” include those that result in mutations due to random errors in DNA replication whereas “extrinsic factors” are environmental factors that affect mutagenesis rates (such as UV radiation, ionizing radiation, and carcinogens). For example, radiation can cause DNA damage, which would primarily result in deleterious mutations with functional consequences on cancer development only after cell division. Therefore, extrinsic factors may act through the accumulation of genetic alterations during cell division to increase cancer risk. Accordingly, intrinsic risk would result from those apparently uncontrollable intrinsic processes (Arrow 1, Figure below) as well as from those highly modifiable and thus preventable extrinsic factors (Arrow 2, Figure below).
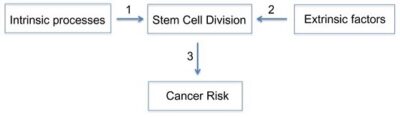
Figure above shows schematic view of how intrinsic processes and extrinsic factors are related to cancer risks through stem-cell division.
______
______
Carcinogen:
A carcinogen is any substance, radionuclide, or radiation that promotes carcinogenesis (the formation of cancer). This may be due to the ability to damage the genome or to the disruption of cellular metabolic processes. Several radioactive substances are considered carcinogens, but their carcinogenic activity is attributed to the radiation, for example gamma rays and alpha particles, which they emit. Common examples of non-radioactive carcinogens are inhaled asbestos, certain dioxins, and tobacco smoke. Although the public generally associates carcinogenicity with synthetic chemicals, it is equally likely to arise from both natural and synthetic substances. Carcinogens are not necessarily immediately toxic; thus, their effect can be insidious.
_
Carcinogens are agents in the environment capable of contributing to cancer growth. Carcinogens can be categorized into two different types: activation-dependent and activation-independent, and each nature impacts their level and type of influence when it comes to promoting cancer growth. Activation-dependent carcinogens require metabolic activation or modification to induce cancer, while activation-independents ones do not. Examples of activation-dependent carcinogens range from certain viruses, such as HPV, to consumed alcohol, to excessive amounts of red and processed meats, impacting a person’s health in ways they may not immediately associate with cancer. Activation-independent carcinogens, such as ultraviolet rays or nitrosamines in tobacco products, possess characteristics enabling them to interact directly with DNA and other cellular components to cause harm. These include not requiring metabolic action or molecular changes to act, which complements their ability to be electrically excited, permitting them to interact with oxygen and nitrogen atoms in negatively charged cellular environments. This type of interaction leads to the alteration of DNA nucleotide bases, causing disarrangement of that genetic material. This disarrangement is also responsible for the formation of DNA adducts, segments of DNA which bind to carcinogens, which furthers harm. Eventually, failure in DNA repair mechanisms will lead to a build-up of DNA damage and potentially the development of cancer.

_
Cancer is any disease in which normal cells are damaged and do not undergo programmed cell death as fast as they divide via mitosis. Carcinogens may increase the risk of cancer by altering cellular metabolism or damaging DNA directly in cells, which interferes with biological processes, and induces the uncontrolled, malignant division, ultimately leading to the formation of tumors. Usually, severe DNA damage leads to programmed cell death, but if the programmed cell death pathway is damaged, then the cell cannot prevent itself from becoming a cancer cell.
_
There are many natural carcinogens. Aflatoxin B1, which is produced by the fungus Aspergillus flavus growing on stored grains, nuts and peanut butter, is an example of a potent, naturally occurring microbial carcinogen. Certain viruses such as hepatitis B and human papilloma virus have been found to cause cancer in humans. The first one shown to cause cancer in animals is Rous sarcoma virus, discovered in 1910 by Peyton Rous. Other infectious organisms which cause cancer in humans include some bacteria (e.g. Helicobacter pylori) and helminths (e.g. Opisthorchis viverrini and Clonorchis sinensis). Dioxins and dioxin-like compounds, benzene, kepone, EDB, and asbestos have all been classified as carcinogenic. As far back as the 1930s, industrial smoke and tobacco smoke were identified as sources of dozens of carcinogens, including benzo[a]pyrene, tobacco-specific nitrosamines such as nitrosonornicotine, and reactive aldehydes such as formaldehyde, which is also a hazard in embalming and making plastics. Vinyl chloride, from which PVC is manufactured, is a carcinogen and thus a hazard in PVC production.
_
Mechanisms of carcinogenicity:
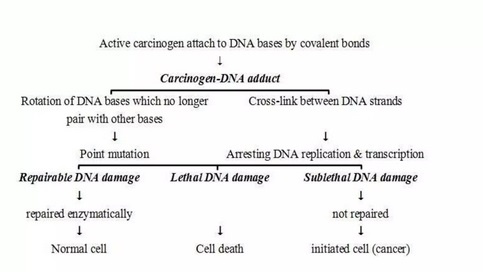
Carcinogens can be classified as genotoxic or nongenotoxic. Genotoxins cause irreversible genetic damage or mutations by binding to DNA. Genotoxins include chemical agents like N-nitroso-N-methylurea (NMU) or non-chemical agents such as ultraviolet light and ionizing radiation. Certain viruses can also act as carcinogens by interacting with DNA. A nucleophile is usually negatively charged or neutral with a lone pair of electrons. H2O, -OMe or -OtBu are some examples. Overall, the electron-rich species is a nucleophile. Electrophiles are generally positively charged or neutral species with empty orbitals attracted to a centre rich in electrons. It is believed that the most nucleophilic atoms of purines are the N7 atom of guanine and N1 atom of adenine on DNA. DNA is nucleophilic; therefore, soluble carbon electrophiles are carcinogenic, because DNA attacks them. For example, some alkenes are toxicated by human enzymes to produce an electrophilic epoxide. DNA attacks the epoxide, and is bound permanently to it. This is the mechanism behind the carcinogenicity of benzo[a]pyrene in tobacco smoke, other aromatics, aflatoxin and mustard gas.
Nongenotoxins do not directly affect DNA but act in other ways to promote growth. These include hormones and some organic compounds.
_____
Carcinogenic Agents:
Although a clearly defined cause is not apparent in the majority of de novo malignancies, it is well documented that carcinogenic agents contribute to many human cancers. These fall into several broad groups, which can be categorized as infective (e.g., viruses), chemical (including occupational, environmental, and therapeutic), electromagnetic radiation (i.e., ultraviolet [UV], X-ray, gamma ray), and immunosuppressive (HIV, immunosuppressive medications). These are shown in Table below. Several hundred chemicals have been shown to be carcinogenic in humans. Harm from these chemical carcinogens can be a result of occupational (asbestos, aniline dyes), environmental (alcohol, tobacco), or iatrogenic (chemotherapy) exposure. While many carcinogens are directly mutagenic to DNA, most carcinogens undergo activation after exposure to reactive metabolites that are responsible for genetic damage.
Of the known environmental carcinogens, perhaps the most well-documented agent known are in Table below:
|
Chemicals |
Radiation |
Infectious |
Dietary |
Iatrogenic |
|
Occupational |
UV light |
Virus |
Fat |
Radiation |
|
Arsenic |
X-ray |
HPV |
Calorie |
Immunosuppression |
|
Asbestos |
Gamma ray |
EBV |
Low fiber |
Chemotherapy |
|
Coal tars |
Nuclear radiation |
KSHV |
||
|
Soot |
HTLV-1 HEP B/C |
|||
|
Benzene |
||||
|
Vinyl chloride |
||||
|
Behavioral |
Bacteria |
|||
|
H. pylori C. psittaci |
||||
|
Tobacco |
Fungus |
|||
|
A. flavus |
||||
|
Alcohol |
Parasite Schistosoma |
|||
|
Clonorchis |
______
Identifying carcinogens:
The IARC Monographs identify the causes of human cancer, based on the systematic assembly, review, and integration of evidence of cancer in humans, cancer in experimental animals, and carcinogen mechanisms. Of the approximately 120 agents classified by the IARC Monographs as carcinogenic to humans (Group 1), most have sufficient evidence of carcinogenicity in humans, based on epidemiological studies. However, epidemiological studies of cancer in exposed humans are often limited in number, and may have deficiencies in terms of sample size, confounding, and exposure characterization. Furthermore, for chemicals that have recently been introduced on the market, epidemiological studies may not exist or may not be relevant, because of the long latency period for cancer development. The number of lifetime rodent cancer bio- assays being performed is declining, and only a fraction of the approximately 75000 chemicals that are listed in the Toxic Substances Control Act Chemical Substance Inventory of the United States Environmental Protection Agency (EPA) have been formally evaluated by the United States National Toxicology Program (NTP) or other national testing programmes (e.g. the Japan Bioassay Research Center of the Japan Organization of Occupational Health and Safety). In contrast, data on carcinogen mechanisms from human biomarker studies, in vivo animal tests, and in vitro cell culture models are increasing in both volume and diversity.
_
Agents Classified by the IARC Monographs, Volumes 1–133:
|
Group 1 |
Carcinogenic to humans |
126 agents |
|
Group 2A |
Probably carcinogenic to humans |
94 agents |
|
Group 2B |
Possibly carcinogenic to humans |
322 agents |
|
Group 3 |
Not classifiable as to its carcinogenicity to humans |
500 agents |
_
When the evidence from human epidemiological studies is less than sufficient, strong mechanistic data can play a pivotal role in the overall carcinogen hazard classification. For instance, even though the evidence from rodent cancer bioassays provided sufficient evidence of carcinogenicity in experimental animals, d-limonene was categorized as not classifiable as to its carcinogenicity to humans (Group 3) on the basis of mechanistic and other relevant data, because the probable mechanism of carcinogenicity in experimental animals was unlikely to operate in humans. Other agents have been classified as probably carcinogenic to humans (Group 2A) or even as carcinogenic to humans (Group 1) based on strong evidence for recognized carcinogen mechanisms, such as genotoxicity (for ethylene oxide), inhibiting DNA repair (for etoposide), or binding to the aryl hydrocarbon receptor and subsequent downstream effects (for 2,3,7,8-tetrachlorodibenzo-para-dioxin).
_
The key characteristics of human carcinogens were recently introduced as the basis for a uniform approach to evaluating mechanistic evidence to support cancer hazard identification. The key characteristics reflect the chemical and biological properties of established human carcinogens, including “is genotoxic”, “is immunosuppressive”, and “modulates receptor-mediated effects”. The key characteristics are distinct from the hallmarks of cancer, which relate to the properties of cancer cells. The key characteristics approach avoids a narrow focus on specific pathways and hypotheses and provides for a broad, holistic consideration of the mechanistic evidence. Therefore, data on the key characteristics can provide independent evidence of carcinogenicity when data from studies in humans are lacking, and can help in establishing biological plausibility. The key characteristics approach is being increasingly applied by agencies throughout the world, and key characteristics for other toxicological hazards are being developed. The key characteristics approach can inform the design of high-throughput testing systems and human biomarker studies with greater relevance to cancer hazard identification – the first step in cancer prevention. The recent description of certain key characteristics, one or more of which is exhibited by all established human carcinogens, is an innovative approach to identifying carcinogens. The number of ways in which agents contribute to carcinogenesis can be extensive. However, these mechanisms can be grouped into a limited number of categories (genotoxicity, immunosuppression, etc.).
_
Key characteristics of carcinogens:
-1. Is electrophilic or can be metabolically activated to electrophiles
-2. Is genotoxic
-3. Alters DNA repair or causes genomic instability
-4. Induces epigenetic alterations
-5. Induces oxidative stress
-6. Induces chronic inflammation
-7. Is immunosuppressive
-8. Modulates receptor-mediated effects
-9. Causes immortalization
-10. Alters cell proliferation, cell death, or nutrient supply
_
Characteristic 1: Is electrophilic or can be metabolically activated to electrophiles
Electrophiles are electron-seeking molecules that form addition products, commonly referred to as adducts, with cellular macromolecules including DNA, RNA, lipids, and proteins. Some chemical carcinogens (e.g. sulfur mustard) are direct-acting electrophiles, whereas others (e.g. aflatoxins, benzene) require chemical conversion within the body or metabolic activation. The ability to form adducts with nucleic acids and proteins is a common property of these inherently electrophilic and/or metabolically activated human carcinogens.
Characteristic 2: Is genotoxic
A genotoxic agent induces damage to a cell’s genetic material. Examples of DNA damage include DNA strand breaks (breaks in the phosphodiester bonds), protein–DNA cross-links, and oxidative damage to DNA. Genotoxic agents may also induce damage at the chromosomal level, including chromosomal aberrations, micronuclei, sister chromatid exchanges, and aneuploidy. A mutation, which is a change in the DNA sequence, usually arises as the cell attempts to repair the DNA damage. A large proportion of the agents classified by IARC in Group 1 are genotoxic.
Characteristic 3: Alters DNA repair or causes genomic instability
Carcinogens may act not only by producing DNA damage directly but also by altering the processes that control normal DNA replication or repair of DNA damage. Examples include the inhibition of DNA repair by cadmium and formaldehyde. In cells exposed to ionizing radiation, genetic instability is a relatively late-occurring event that appears several cell generations after irradiation and results in a reduced ability to replicate the genotype faithfully.
Characteristic 4: Induces epigenetic alterations
The term “epigenetic” refers to stable changes in gene expression and chromatin organization that are not caused by changes in the DNA sequence itself and can be inherited over cell divisions. Epigenetic phenomena – including changes in the DNA methylome, in chromatin compaction states, and in histone modification – are important aspects of normal developmental processes that can be usurped during the carcinogenic process, with impacts on gene expression and DNA repair dynamics. A wide range of carcinogens have been shown to dysregulate the epigenome.
Characteristic 5: Induces oxidative stress
Many carcinogens are capable of influencing redox balance within target cells. If an imbalance occurs, favouring the formation of reactive oxygen species at the expense of their detoxification, this is referred to as oxidative stress. This may be accompanied by the production of reactive nitrogen species, or nitrative stress. Oxidative stress can lead to the generation of mutations in DNA, and more than 100 different types of oxidative damage to DNA have been identified. The induction of oxidative stress and subsequent injury is a characteristic of a diverse group of carcinogens, including radiation, asbestos, chemicals, and carcinogenic infectious agents.
Characteristic 6: Induces chronic inflammation
Chronic inflammation from persistent infections, such as that caused by Helicobacter pylori, has been associated with several forms of cancer. Various other carcinogens also induce chronic inflammation, including fibres (e.g. silica, asbestos) and chemicals (e.g. polychlorinated biphenyls).
Characteristic 7: Is immunosuppressive
Immunosuppression is a reduction in the capacity of the immune system to respond effectively to foreign antigens, including antigens on tumour cells. Persistent immunosuppression presents a risk of cancer, especially excess risk of lymphoma. Several carcinogens act entirely or largely by immunosuppression, often in concert with oncogenic infectious agents. The Group 1 agents that act by immunosuppression include HIV-1 and the immunosuppressive drug ciclosporin (also known as cyclosporine).
Characteristic 8: Modulates receptor-mediated effects
All actions of hormonally active agents are mediated by their ability to interact with a receptor, with the hormone acting as an endogenous ligand. For a chemical to interfere with hormone signalling and produce adverse effects, it must ultimately interfere with hormone receptor activation – either directly or indirectly. Numerous carcinogens act as ligands to receptor proteins, including hormone replacement therapy and 2,3,7,8-tetrachlorodibenzo- para-dioxin. Many exogenous agents act directly as agonists or antagonists by competing for binding with the endogenous ligand (e.g. a hormone, such as testosterone). However, there are also receptors for which few or no endogenous ligands have been identified, such as the aryl hydrocarbon receptor; in these cases, the carcinogenic chemical is the activating ligand. Carcinogens may also act indirectly on receptor-mediated effects by altering the bioavailability of endogenous ligands by affecting the biosynthesis, bioactivation, and/ or degradation of the ligand. These direct and indirect effects all modulate receptor-based regulation of gene transcription, and ultimately cell growth and proliferation.
Characteristic 9: Causes immortalization
Several human DNA and RNA viruses are carcinogenic to humans. Although oncogenic viruses belong to different families, their strategies in human cancer development show many similarities and involve viral encoded oncoproteins targeting the key cellular proteins that regulate cell growth. These targets may include important tumour suppressor genes and/or oncogenes. The result of these viral effects is to immortalize the cells of the target tissue such that they divide continuously.
Characteristic 10: Alters cell proliferation, cell death, or nutrient supply
A component common to many types of cancer is the evasion of programmed cell death, via apoptosis, or of other terminal programming, including autophagy, in at least a proportion of the cell population. In contrast to apoptosis and autophagy, necrotic cell death releases pro-inflammatory signals into the surrounding tissue, which can enhance cancer cell proliferation and promote cancer metastasis. Many agents affect necrosis, apoptosis, and/or autophagy, and they can have profoundly divergent effects on cancer induction in different tissues. In addition to cell death caused directly by the toxicity of an agent, cells within a tumour may die as a result of an impaired nutrient supply. The number of neoplastic cells can increase exponentially, quickly outstripping the supply capabilities of the existing tissue vasculature. Neo-angiogenesis, in which new blood vessels grow into a tumour, is key to providing a supply of nutrients. Thus, agents that promote angiogenesis, such as arsenic, will promote tumour growth.
_____
_____
Free radicals and cancer:
Free radicals are highly reactive chemicals that have the potential to harm cells. A free radical is atom or group of atoms that has at least one unpaired electron and is therefore unstable and highly reactive (often only occur as transient species). Free radicals are formed naturally in the body and play an important role in many normal cellular processes. At high concentrations, however, free radicals can be hazardous to the body and damage all major components of cells, including DNA, proteins, and cell membranes. The damage to cells caused by free radicals, especially the damage to DNA, may play a role in the development of cancer and other health conditions. Free radicals that contain the element oxygen are the most common type of free radicals produced in living tissue. Another name for them is “reactive oxygen species,” or “ROS”.
Abnormally high concentrations of free radicals in the body can be caused by exposure to ionizing radiation and other environmental toxins. When ionizing radiation hits an atom or a molecule in a cell, an electron may be lost, leading to the formation of a free radical. The production of abnormally high levels of free radicals is the mechanism by which ionizing radiation kills cells. Moreover, some environmental toxins, such as cigarette smoke, some metals, and high-oxygen atmospheres, may contain large amounts of free radicals or stimulate the body’s cells to produce more free radicals.
Antioxidants are chemicals that interact with and neutralize free radicals, thus preventing them from causing damage. Antioxidants are also known as “free radical scavengers.” The body makes some of the antioxidants that it uses to neutralize free radicals. These antioxidants are called endogenous antioxidants. However, the body relies on external (exogenous) sources, primarily the diet, to obtain the rest of the antioxidants it needs. These exogenous antioxidants are commonly called dietary antioxidants. Fruits, vegetables, and grains are rich sources of dietary antioxidants. Some dietary antioxidants are also available as dietary supplements. Examples of dietary antioxidants include beta-carotene, lycopene, and vitamins A, C, and E (alpha-tocopherol). The mineral element selenium is often thought to be a dietary antioxidant, but the antioxidant effects of selenium are most likely due to the antioxidant activity of proteins that have this element as an essential component (i.e., selenium-containing proteins), and not to selenium itself.
_
Genetic polymorphisms of cellular antioxidants are known to play a significant role in the pathogenesis of various oxidative stress- and inflammation-related diseases, such as cancer. Recently, a meta-analysis study has stated that catalase C262 T polymorphism is associated with an increased risk of prostate cancer. Similarly, polymorphisms in SOD gene is associated with the onset of different cancer types, including lung and colorectal cancers. In case of lung cancer, one study has shown that glutathione S-transferase T1 gene polymorphism is associated with the lung cancer risk. Similarly, manganese SOD (MnSOD) gene polymorphisms (Ala16Val) together with the smoking status are known to be associated with an increased risk of lung cancer.
_
Can antioxidant supplements help prevent cancer?
In laboratory and animal studies, the presence of increased levels of exogenous antioxidants has been shown to prevent the types of free radical damage that have been associated with cancer development. Therefore, researchers have investigated whether taking dietary antioxidant supplements can help lower the risk of developing or dying from cancer in humans. Many observational studies, including case–control studies and cohort studies, have been conducted to investigate whether the use of dietary antioxidant supplements is associated with reduced risks of cancer in humans. Overall, these studies have yielded mixed results. Because observational studies cannot adequately control for biases that might influence study outcomes, the results of any individual observational study must be viewed with caution. Randomized controlled clinical trials, however, lack most of the biases that limit the reliability of observational studies. Therefore, randomized trials are considered to provide the strongest and most reliable evidence of the benefit and/or harm of a health-related intervention. To date, nine randomized controlled trials of dietary antioxidant supplements for cancer prevention have been conducted worldwide. Many of the trials were sponsored by the National Cancer Institute. Overall, these nine randomized controlled clinical trials did not provide evidence that dietary antioxidant supplements are beneficial in primary cancer prevention. In addition, a systematic review of the available evidence regarding the use of vitamin and mineral supplements for the prevention of chronic diseases, including cancer, conducted for the United States Preventive Services Task Force (USPSTF) likewise found no clear evidence of benefit in preventing cancer.
_____
_____
Cancer-related inflammatory conditions:
Since 1863, when Virchow first hypothesized that cancer develops as the product of unresolved inflammation (Balkwill and Mantovani 2001), tumor-associated inflammation has been key to shaping our modern understanding of cancer progression as seen in the figure below:
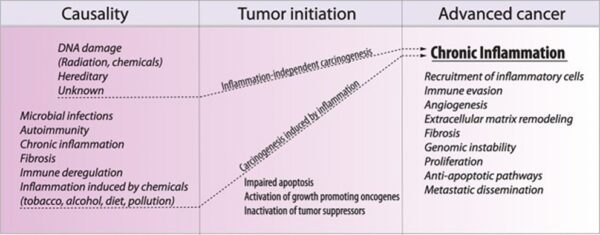
Today, it is accepted that chronic inflammation is a critical hallmark of cancer, with at least 25% of cancers associated with it (Hussain et al. 2000; Coussens and Werb 2002; Beaugerie et al. 2013), and possible underlying causes include microbial infections, autoimmunity, and immune deregulation. For example, human papilloma viruses (HPVs) induce inflammation and are responsible for 90%–100% of all cervical cancers (Bosch et al. 2002). Similarly, chronic infection with Helicobacter pylori elevates the risk for gastric cancer (Hussain et al. 2000). In addition, the immune deregulation seen in inflammatory bowel disease increases colorectal cancer incidence (Lakatos and Lakatos 2008). The nonhuman form of sialic acid—N-glycolylneuroaminic acid (Neu5Gc)—in red meat can be incorporated into human tissue and recruit inflammatory cells (Samraj et al. 2015). In this sense, diet may play a causal role in induction of cancer-associated inflammation (Gupta et al. 2010). Importantly, tobacco and obesity, both of which induce low-grade inflammation, give rise to elevated risks of cancer (Howe et al. 2013); this evidence suggests that the majority of cancers is associated with unresolved inflammation.
_
A connection between inflammation and cancer has long been perceived. Inflammatory cells including macrophages, neutrophils, mast cells, and eosinophils are present in the TME. Tumour-associated macrophages (TAMs) are prototypic inflammatory cells, playing a key role in the orchestration of the TME.
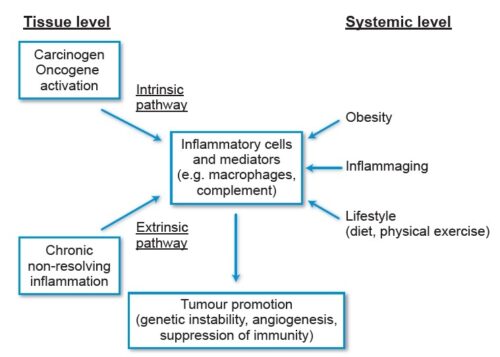
Figure above shows pathways connecting inflammation and cancer at the tissue level and at the systemic level.
_
Chronic inflammation is a necessary consequence of cancer progression. Different inflammatory conditions can lead to neoplastic transformation. However, whether or not the inflammation is present in the origin of carcinogenesis, most tumors progress to a state of chronic inflammation that fuels different aspects of tumor progression, including genomic and epigenomic instability, immune evasion, angiogenesis, and metastatic dissemination.
While chronic inflammation has an important role in cancer, less is known about the impact of acute inflammation on tumor progression. For example, inducing acute inflammation locally in the bladder with a vaccine containing an attenuated Mycobacterium bovis strain successfully treats squamous cancer of the bladder (Askeland et al. 2012). Hence, with the infiltration of leukocytes and subsequent inflammation, the impact from inflammatory mediators can both initiate and, in certain cases, eliminate tumor cells and prevent tumor development (Shalapour and Karin 2015). This dual role of inflammation also becomes evident in the clinic, where immunodeficient patients are more often diagnosed with cancer (Frisch et al. 2001). Interestingly, long-term use of nonsteroidal anti-inflammatory drugs (NSAIDs), which suppresses the immune system, has been linked to a lower risk of cancer (Thun et al. 2002).
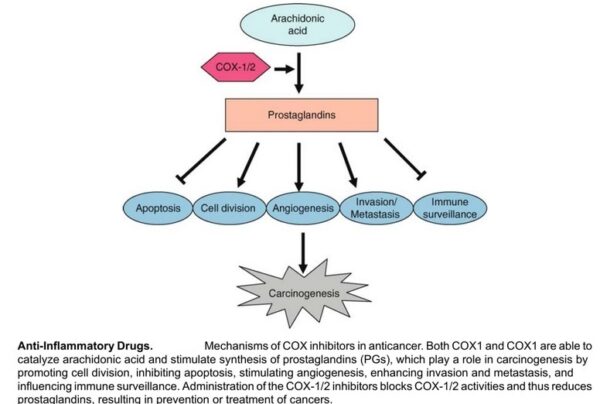
Figure above shows multiple mechanisms of NSAIDs lowering cancer risk.
Studies highlighting cancer protecting effects of NSAIDs:
-1. Nonsteroidal Anti-inflammatory Drugs and Cyclooxygenase-2 Inhibitors for Primary Prevention of Colorectal Cancer: A Systematic Review Prepared for the U.S. Preventive Services Task Force 2007:
Cyclooxygenase-2 inhibitors and NSAIDs reduce the incidence of colonic adenomas. Nonsteroidal anti-inflammatory drugs also reduce the incidence of colorectal cancer (CRC). However, these agents are associated with important cardiovascular events and gastrointestinal harms. The balance of benefits to risk does not favor chemoprevention in average-risk individuals.
-2. Chemoprevention of colorectal cancer: systematic review and economic evaluation, 2010:
Study found that aspirin, celecoxib and calcium may reduce recurrence of adenomas in individuals with an increased risk of colorectal cancer. Chemoprevention may be a cost-effective intervention when targeted at intermediate-risk populations following polypectomy.
-3. Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials, 2011 study:
Daily aspirin reduced deaths due to several common cancers during and after the trials. Benefit increased with duration of treatment and was consistent across the different study populations. These findings have implications for guidelines on use of aspirin and for understanding of carcinogenesis and its susceptibility to drug intervention.
_
Whether or not inflammation is a cause or a consequence, the tumor microenvironment (TME) is compromised, triggering an immune inflammatory response, and histopathological analyses provide evidence for the presence of innate and adaptive immune cells in most human tumors, which are characterized as features of cancer progression (Fridman et al. 2012). There is evidence that inflammation itself plays an important role in the development and progression of cancer. Chronic inflammation can lead to DNA damage over time and the accumulation of random genetic alterations in cancer cells. Inflammation can contribute to proliferation, survival, angiogenesis and migration of cancer cells by influencing tumor microenvironment. Individuals with inflammatory bowel disease are at increased risk of developing colorectal cancers.
_____
IBD and cancer:
Crohn’s disease (CD) and ulcerative colitis (UC) are chronic inflammatory conditions of the gastrointestinal tract. Although the disease pathogenesis is not fully understood, inflammatory bowel disease (IBD) is characterized by chronic inflammation of the gastrointestinal tract in genetically susceptible individuals exposed to environmental risk factors. Together, IBD is estimated to affect more than 0.4% of Europeans and North Americans, a number that is expected to increase over time. Patients with IBD are at an increased risk of cancer secondary to long-standing intestinal inflammation and secondary to immunosuppressive therapies. As the population of patients with IBD ages, there is an increasing risk of cancer development. It is well recognized that patients with IBD are at an increased risk of developing colorectal cancer (CRC), primarily the result of chronic intestinal inflammation. More recently, patients with IBD have also been shown to be at increased risk of developing extra-intestinal malignancies, thought to be a consequence of immunosuppressive therapies and an underlying inflammatory state.
_
Cancer secondary to chronic intestinal inflammation:
In patients with IBD, chronic intestinal inflammation is the primary risk factor for the development of gastrointestinal malignancy. Cancers as a result of chronic intestinal inflammation include CRC, small bowel adenocarcinoma, intestinal lymphoma, anal cancer and cholangiocarcinoma as seen in the table below:
Cancer secondary to chronic intestinal inflammation:
|
Cancer type |
Standardized incidence ratio |
|
Colorectal cancer |
5.7 (95%CI: 4.6-7.0) |
|
Small bowel adenocarcinoma |
27.1 (95%CI: 14.9-49.2) |
|
Intestinal lymphoma |
17.51 (95%CI: 6.43-38.11) |
|
Anal cancer |
Data not available |
|
Cholangiocarcinoma |
916.63 (95%CI: 297.88-2140.99) in UC |
UC: Ulcerative colitis.
_
The risk and pathogenesis of inflammation-associated cancer has chiefly been described in colitis-associated CRC. In a meta analysis, quantitative estimates of CRC risk in UC have been reported to be 2% after 10 years, 8% after 20 years, and 18% after 30 years of disease. Moreover, studies of CRC in UC have noted a high concordance between CRC risk with the location and extent of disease, with a standardized incidence ratio (SIR) of 1.7 for proctitis, 2.8 for left-sided colitis, and 14.8 for pancolitis. All of these studies support the strong association between inflammation and cancer development.
_
Patients with IBD develop colon cancer in a manner similar to well described sporadic molecular mechanisms including mutations in the adenomatous polyposis coli (APC) gene, aneuploidy, DNA methylation, microsatellite instability (MSI), activation of the oncogene k-ras, activation of COX-2, and mutation in tumor suppressor genes DCC/DPC4, and eventual loss of p53 function. However, underlying colonic inflammation changes the timing and sequence of these genomic changes, yielding a process of carcinogenesis that is faster and multifocal. Contrary to sporadic cancers in which the dysplastic precursor is the adenomatous polyp, dysplasia in patients with IBD can be localized, diffuse, or multifocal.
Studies mapping genomic instability secondary to DNA aneuploidy in patients with IBD indicate that these cell populations became more widely distributed, occupying larger areas of colonic mucosa. Over time, further subpopulations with increasingly unstable genomics arise and expand, representing a whole field change, marking the entire colon at risk for further carcinogenesis.
_
Cancer secondary to immunosuppression:
Given that chronic inflammation underlies the disease state of IBD, medications that mitigate inflammation by suppression of the immune system represent the cornerstone of treatment. In addition to treating IBD, it is postulated that these medications, such as immunomodulators [thiopurines (azathioprine or mercaptopurine) or methotrexate] and biologic agents (TNF-α antagonists), may reduce the incidence of inflammation-associated cancer. However, given that immunomodulators and biologic agents act on the immune system, they may also promote carcinogenesis.
Thiopurines and methotrexate promote the development of cancer by a variety of mechanisms including direct alteration in DNA, activation of oncogenes, reduction in physiologic immunosurveillance of malignant cells, and impaired immune control of oncogenic viruses. Less is known about the carcinogenic potential of biologic therapies that block TNF-α and existing molecular data is inconsistent. TNF-α has been shown to exhibit anti-tumor effects by initiating cellular apoptosis of malignant cells, but it is secreted by most tumors to facilitate cellular survival and enhance neoplastic proliferation as a pro-tumor inflammatory cytokine.
Several studies have indicated a risk of therapy-associated malignancies in IBD patients. Population-based cohort and meta-analyses have demonstrated that current use of thiopurines for IBD is associated with a 1.3 to 1.7 overall relative risk of cancer, which is reversible after withdrawal. Current exposure to TNF-α antagonists has not been shown to be associated with an overall excess risk of cancer, but data is very limited. Specific cancers thought secondary to long-standing immunosuppression in the setting of IBD include lymphomas, acute myeloid leukemia, myelodysplastic syndromes, skin cancers, and urinary tract cancers as seen in the table below:
Cancer secondary to immunosuppression:
|
Increased risk under anti-metabolites |
Increased risk under anti-TNFα |
Increased risk under anti-metabolite with anti-TNFα |
|
Non-Hodgkin lymphoma |
Melanoma |
Hepatosplenic T-cell lymphoma |
|
Acute myeloid leukemia and Myelodysplastic syndromes |
||
|
Non-melanoma skin cancers (basal and squamous cell carcinomas) |
||
|
Urinary tract cancers |
TNF-α: Tumor necrosis factor alpha.
_____
_____
Mechanical Trauma causing cancer:
The normal wear and tear of life induces a multiplicity of traumas which are rarely noted or quickly forgotten until the time arises to make something out of them. The role of trauma in the causation of cancer is a subject fraught with gross exaggerations and contradictions. The literature abounds in points of view ranging from detailed descriptions of case reports of trauma followed by malignant neoplasms to others in which this relationship is minimized or flatly denied.
A wide variety of malignant neoplasms have been described in association with trauma. In many cases, the association appears purely coincidental, judging from the lack of scientific evidence. Mechanical trauma may soon alert the patient to the presence of a pre-existing neoplasm in the affected part. Different forms of skin cancer have been known to result from mechanical injury.
In one of the most important epidemiologic studies relating trauma and tumor incidence, Inskip and colleagues studied, over a 15-year period, 228,055 Danish residents who had been hospitalized for head injury and observed for at least 5 years, an average of 8, and a maximum of 17 years for benign and malignant brain tumors. Because of the superb Danish identification techniques, a search of the national tumor registry was conducted for the same 15-year period plus 1 year, and the results compared with the incidence in the Danish population. Excluding the first year of follow-up, which probably was heavily weighted by prevalent cases present before the injury, the standardized incidence ratio was 1.0 for gliomas, and 1.2 for meningiomas (with confidence intervals [CI] of 0.8 to 1.7). Thus, no significant relationship to trauma was found for the two most common types of brain tumor. Hemangioma and hemangioblastoma (N=15) were more frequent than in the general population, for a standardized incidence ratio of 2.6 (CI 1.4–4.2). This correlation needs independent confirmation.
A multi-region study in the United Kingdom identified 794 men aged 15 to 49 years diagnosed with testicular germ cell tumor in a 33-month period. An age-matched control subject was selected, and all were interviewed concerning testicular trauma at least 2 years prior to the tumor. The odds ratio was 2.00 (CI 1.54–2.61). Although this difference is significant, it depends on memory of preceding events, which may have been enhanced by the subsequent attention to the testis.
_
There is no experimental evidence to substantiate the production of tumors by the direct action of trauma, but there are numerous studies indicating its significance as a “promoter.” The studies of Rous and co-workers showed that under certain circumstances, in rabbits, a trauma can precipitate the formation of a tumor when the cells have been conditioned with previous oncogenic tar treatment. If trauma is to be considered independently as a tumor-causative factor, however, this could only take place as an extremely rare event during the process of regeneration and repair. A normal regenerative process implies the restoration of lost tissues by structurally and often functionally similar cells. A variety of organ-specific wound hormones (cytokines) liberated from the site of injury exert a stimulating effect on homologous tissue. Such putative cytokine release might eliminate or decrease an inhibitory gene regulatory effect, allowing increased cell cycling and growth. On purely numerical grounds, an increased number of mutations might then ensue. It is conceivable that persistent or repetitive cell damage by trauma could trigger an excessive proliferative effect. This is quite different from a single traumatic event, however.
The possibility of direct mutation from chronic inflammation or repair is controversial. Although this is the presumed mechanism of epithelial carcinogenesis in burn cancers and long-draining sinuses (although many cytokines may be involved) the rarity of soft tissue sarcomas compared with the frequency of wounding, operative and blunt trauma to mesenchymal tissues casts doubt on traumatic mutation in these tissues as an oncogenic event.
_____
_____
Cancer in Dialysis and Transplant Patients:
The etiology of cancer risk in dialysis patients includes the presence of chronic infection (especially in the urinary tract), a depressed immune system, previous treatment with immunosuppressive or cytotoxic drugs, nutritional deficiencies, and altered deoxyribonucleic acid (DNA) repair mechanisms. Importantly, cancer is not related to dialysis modality, but rather the uremic state. Uremia is associated with impaired T cell immunity and a state of chronic inflammation, which lead to DNA mutations in proliferating cells and deregulatory release of cytokines implicated in cancer development and progression. CKD also leads to the accumulation of carcinogenic compounds to which dialysis patients are exposed from the environment and possibly in the dialysate. Increasing the frequency of dialysis has been associated with reduced genomic damage and plasma urea concentrations in patients with ESKD. Excess cancer risk may also be due to an interaction of immune dysfunction induced by uremia with established risk factors (ultraviolet [UV] radiation, tobacco, alcohol). Several reports have also demonstrated high levels of cumulative radiation dose in patients with ESKD on dialysis. Although there have been no follow-up studies that have measured cumulative radiation doses and cancer outcomes in patients with ESKD, high exposure (cumulative effective dose >50 mSv) has been reported to increase cancer mortality by 5% in other populations.
In addition to the persistent metabolic changes associated with ESKD, the underlying causative disease, and the development of certain complications of ESKD may also predispose to cancer. The risk of renal cell cancer (RCC) is increased in patients with acquired cystic disease and seems to be related to the total duration of CKD, rather than the duration of dialysis. Other conditions predisposing to cancer include Balkan nephropathy and analgesic nephropathy, both of which are associated with a high risk of developing tumors of the renal pelvis and ureter.
The increased risk of some cancer types is rapidly reversed when immunosuppression is reduced or withdrawn after kidney transplant failure. These cancer types include Kaposi’s sarcoma, non-Hodgkin lymphoma, melanoma, and squamous cell carcinomas of the lip. However, the risk of cancer at other sites remains significantly elevated after iatrogenic immunosuppression is ceased. These cancer types include leukemia, lung cancer, and cancers related to ESKD.
The frequency of viral infections in dialysis patients is poorly documented, but there is no doubt that patients with ESKD have a greater than normal exposure to hepatitis B and hepatitis C viruses, and this probably accounts for the observed excess of liver cancer. Human papillomavirus (HPV) is associated with cancers of the tongue, cervix, vagina, vulva, and penis. HIV is also associated with increased risk of Kaposi’s sarcoma, non-Hodgkin’s lymphoma, and Hodgkin’s lymphoma, lip, and cervical cancers. In both dialysis patients and transplant recipients, the increased risk of lymphoma is thought likely to be due to activation of dormant Epstein-Barr virus (EBV).
______
______
Occupational Cancer:
Though everyday exposures to chemicals are usually too low to cause harmful health problems, exposure in the workplace can be more serious. Chemical exposures in the workplace can happen at high levels and over long period of time. That is why some jobs require that employees wear protective clothing and equipment and/or respirators.
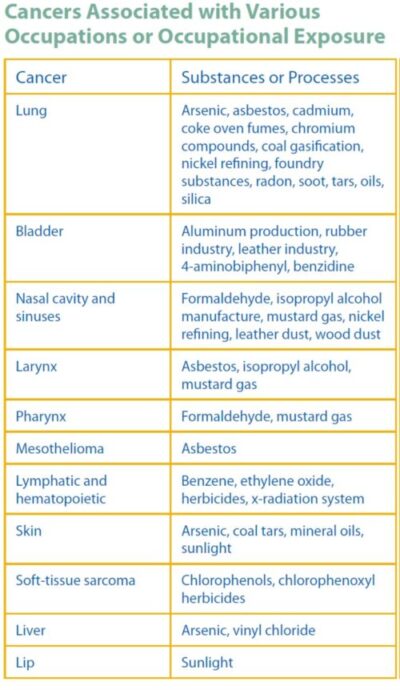
_
The following is a list of substances NIOSH considers to be potential occupational carcinogens:
A number of the carcinogen classifications deal with groups of substances: aniline and homologs, chromates, dintrotoluenes, arsenic and inorganic arsenic compounds, beryllium and beryllium compounds, cadmium compounds, nickel compounds, and crystalline forms of silica. There are also substances of variable or unclear chemical makeup that are considered carcinogens, coal tar pitch volatiles, coke oven emissions, diesel exhaust and environmental tobacco smoke.
Some of the potential carcinogens listed in this index may be re-evaluated by NIOSH as new data become available and the NIOSH recommendations on these carcinogens either as to their status as a potential occupational carcinogen or as to the appropriate recommended exposure limit may change.
_
Past estimates indicate that about 4% of cancer deaths in the U.S. are caused by occupational exposures; currently this is thought to underestimate the true burden of occupational cancer. Many of the studies that reported on the health effects of carcinogens were conducted in manufacturing. These assessments have resulted in the monitoring of and reduction in workplace exposures to carcinogens worldwide, in some cases through the development of protective standards. Exposures to carcinogens in the workplace may not result in cancer until 15-40 years later. Prevention of exposure to newly identified carcinogens is critical in order to achieve reductions in workplace attributable cancer. Based on the National Occupational Mortality Surveillance System (NOMS), U.S. manufacturing workers have increased proportionate mortality to cancer before age 65. To reduce cancer in workers, preventive strategies should be used in manufacturing processes where known and potential carcinogens are used.
_____
_____
Poverty as a Risk Factor in Human Cancers:
A very comprehensive review has been published in the recent issue of Iranian J Public Health, entitled “Environmental Factors Inducing Human Cancers” by Dr Parsa from The National Institutes of Health, Bethesda. The author has discussed the instigating factors of environmental cancer and the way they can seriously disrupt in cellular interactions. He believed that in addition to all of the evidence cited, exposure to the environment is linked to multiple causal factors of various human cancers. Furthermore, in addition to the environmental factors that Dr Parsa has identified in his article, poverty is also is among the greatest health risks and a major carcinogen. Dr. Samuel Broder, who was director of the National Cancer Institute, had suggested that “poverty is a carcinogen,” a cancer-causing agent.
_
In the last 50 years, lung cancer mortality has continued to increase in the lower socioeconomic groups but has started to decrease in more socioeconomically favored groups. The explanation, from an epidemiological point of view, is that smoking (as well as drinking alcohol and other harmful behaviors) is more frequent within the lower socioeconomic groups. Poor people also have several other health related problems due to their poverty. For example, their low socioeconomic status typically means a low intake of garden-fresh fruit and vegetables which is associated with a higher risk of gastro intestinal cancers. In addition, poor people are more vulnerable to environmental factors inducing human cancers as Dr Parsa described such as tobacco use, sunlight and ionizing radiation, alcohol consumption, organic and inorganic chemicals, and infectious micro-organisms.
______
______
Section-5
Cancer and Aging:
There are four fundamental mechanisms leading to biological aging:
-1. Inflammation
-2. Cellular Senescence
-3. Macromolecular dysfunction (e.g., changes in DNA, Telomeres etc)
-4. Stem Cell and Progenitor Dysfunction
These processes interact. One process (like inflammation) can initiate one of the others, like cell senescence, and vice versa.
_
Advancing age is the most important risk factor for cancer overall and for many individual cancer types. The incidence rates for cancer overall climb steadily as age increases, from fewer than 25 cases per 100,000 people in age groups under age 20, to about 350 per 100,000 people among those aged 45–49, to more than 1,000 per 100,000 people in age groups 60 years and older.
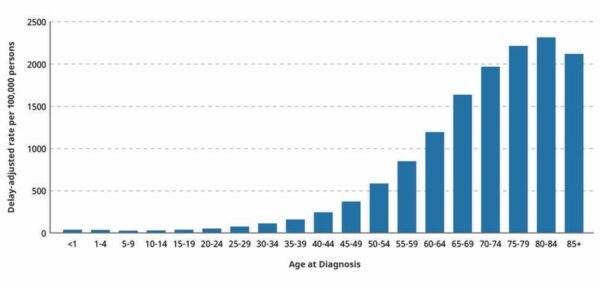
Figure above shows incidence rates by age at diagnosis, all cancer types, all races, and both sexes.
According to the most recent statistical data from NCI’s Surveillance, Epidemiology, and End Results (SEER) Program, the median age of a cancer diagnosis is 66 years. This means that half of cancer cases occur in people below this age and half in people above this age. A similar pattern is seen for many common cancer types. For example, the median age at diagnosis is 62 years for breast cancer, 67 years for colorectal cancer, 71 years for lung cancer, and 66 years for prostate cancer.
But cancer can be diagnosed at any age. For example, bone cancer is most frequently diagnosed in children and adolescents (people under age 20), with about one-fourth of cases occurring in this age group. And 12% of brain and other nervous system cancers are diagnosed in children and adolescents, whereas only 1% of cancer overall is diagnosed in this age group.
_____
Aging is the inevitable time-dependent decline in physiological organ function and is a major risk factor for cancer development. Due to advances in health care, hygiene control and food availability, life expectancy is increasing and the population in most developed countries is shifting to an increasing proportion of people at a cancer susceptible age. Mechanisms of aging are also found to occur in carcinogenesis, albeit with shared or divergent end-results. It is now clear that aging and cancer development either share or diverge in several disease mechanisms. Such mechanisms include the role of genomic instability, telomere attrition, epigenetic changes, loss of proteostasis, decreased nutrient sensing and altered metabolism, but also cellular senescence and stem cell function. Cancer cells and aged cells are also fundamentally opposite, as cancer cells can be thought of as hyperactive cells with advantageous mutations, rapid cell division and increased energy consumption, while aged cells are hypoactive with accumulated disadvantageous mutations, cell division inability and a decreased ability for energy production and consumption. The underlying mechanism in both cancer and aging is the time dependent accumulation of cellular damage. Cancer and aging may seem like opposite processes – cancer cells have the ‘gain of function and fitness’ whereas aging cells are characterized by a ‘loss of function and fitness’. However, the two traits do share many common characteristics as seen in the table below.
Hallmarks that are either shared or divergent in aging and cancer.
|
Hallmarks |
Aging |
Cancer |
|
DNA methylation |
Global hypomethylation |
Hyper- of tumor suppressors and hypo- of oncogenes |
|
Histone modification Non-coding DNA |
Complex |
Complex miRNA deregulation, for example, miR-17-92 upregulation |
|
Proteostasis: |
||
|
Chaperoning |
Impaired |
Augmented |
|
Proteasome activity |
Impaired |
Augmented |
|
Autophagy-lysosome activity |
Impaired |
Augmented |
|
Deregulated nutrient sensing |
Inhibition of insulin and mTOR signaling increase lifespan |
Inhibition of insulin and mTOR signaling is antineoplastic |
|
Cellular senescence |
Increased |
Prevalent in premalignant tumors but evaded in fully malignant tumors |
|
Stem cell |
Exhausted |
Potential nidus for tumorigenesis |
_____
_____
Why advancing age is the most important risk factor for cancer?
As life expectancy has increased dramatically over the past decades, older persons represent a rapidly growing section of our population. This results in an increasing number of older patients with cancer as well. Moreover, as cancer and aging are closely interrelated, cancer incidence is higher in the older age categories compared younger age categories. There are several factors associated with aging that could explain this.
_
-1. Cellular Damage and DNA Damage Response:
Accumulation of cellular damage is probably the most important driver of both aging and cancer, and can be caused by several shared events. Life-long exposure to many endogenous (e.g., free radicals) and exogenous (e.g., UV radiation, foods, etc.) stress factors can induce an increase of oxidative stress, leading to genomic instability and ultimately DNA damage. Reactive oxygen/nitrogen species can react with DNA and can cause several types of DNA damage such as oxidation of purines and pyrimidines, single strand breaks and double-strand breaks. Antioxidant systems like antioxidant enzymes (i.e., catalase and glutathione peroxidase), vitamins (i.e., vitamins C and E) and other radical scavengers (glutathione) are able to prevent oxidative DNA damage. Yet, when this first line defense is ineffective, the DNA damage response is triggered and the cell cycle is arrested to allow DNA repair mechanisms to restore the damage (Figure below). Essential nonredundant players of the DNA damage response are ataxia-telangictasia-mutated (ATM), recruited after a double strand break via the DNA damage sensor complex MRE11-RAD50-NSB1, and ataxia-telangictasia-Rad3-related (ATR), induced after a single strand break detected by RPA, ATRIP or the RAD6-RAD1-HUS1 complex. In their turn, ATM and ATR activate several DNA damage mediators, such as breast cancer gene 1 (BRCA1) and downstream kinases like checkpoint kinases (CHK) 1 and 2. The latter are able to trigger effectors like P53, ensuring initiation of either apoptosis, cell cycle arrest or DNA repair. The two most important mechanisms are base excision repair and nucleotide excision repair. When the DNA repair mechanisms are also unsuccessful, either the apoptotic pathway or senescence program can be activated to eliminate cell carrying potentially dangerous mutations that can lead to cell transformation and tumor initiation.
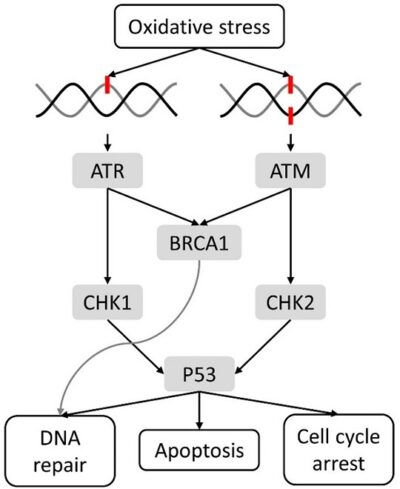
Figure above shows overview of the DNA damage response after oxidative DNA damage. Double strand breaks recruit ataxia-telangictasia-mutated (ATM), whereas single strand breaks induce ataxia-telangictasia-Rad3-related (ATR). DNA damage response mediators and downstream kinase can be activated whereby either DNA repair, apoptosis or cell cycle arrest will occur. Besides direct DNA damage, telomere shortening has been shown to trigger a DNA damage response. Telomere length decreases with aging as telomeres shorten with each cell division. Short telomeres are associated with genomic instability, which occurs in early tumor development, and are a significant risk factor for aging-related diseases.
____
-2. Cellular Senescence:
Cellular senescence is considered to be one of the most important driving forces of the aging process. It is triggered by factors associated with cellular damage, such as oxidative stress, and telomere shortening and expression of oncogenes. The triggers activate several senescence genes, which initiate the actual induction of cellular senescence. The two senescence genes that play critical roles are TP53 and P16INK4a, both are tumor-suppressor genes. The TP53 gene encodes for the P53 protein and is an activator of the cyclin dependent kinase (CDK) inhibitor P21, which will inhibit CDK4/6 activity. The P16INK4a gene makes part of the CDKN2a or INK4a/ARF locus, which based on alternative splicing, encodes two protein products: P14ARF, a regulator of P53 stability and the P16INK4a protein, an inhibitor of CDK4/6. Thus, both P53 and P16INK4a prohibit the activation CDK4/6, thus blocking the phosphorylation of the retinoblastoma protein (pRB). As a result, the cell cycle is arrested in the G1 phase.
In Figure below an overview of the pathways involved in cellular senescence is shown.
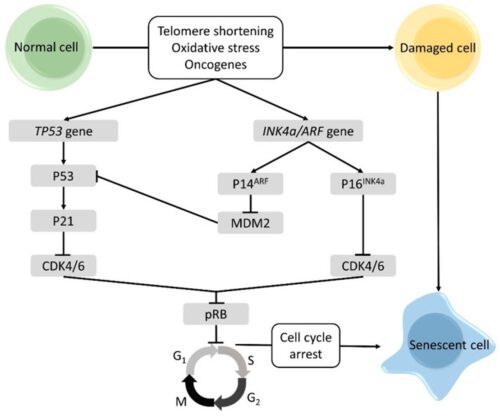
Due to cellular stressors like telomere shortening, DNA damage, oxidative stress and the expression of oncogenes, senescence genes are induced. The TP53 gene encodes for P53, which induces P21, a cyclin dependent kinase inhibitor (CDK) blocking CDK4/6. As a result, phosphorylation of the retinoblastoma protein (pRB) is hindered. Consequently, the cell cannot enter the S-phase and the cell cycle is arrested in the G1 phase. The INK4a/ARF locus encodes for P14ARF and P16INK4a protein. P14ARF is a regulator of P53 activity: by binding mouse double minute 2 homolog (MDM2), degradation of p53 is avoided. Like P53, P16INK4a represses CDK4/6 by which pRB is not phosphorylated and cell cycle progression is prohibited.
_
Senescent cells enter a state of irreversible growth arrest, yet they remain metabolically active. As a result, damaged cells (such as cells having oxidative, DNA damage, shortened telomeres, genomic instability, oncogenic mutations) are unable to proliferate in an uncontrolled manner, which is an important antitumor mechanism. A schematic overview of the antitumor activities of cellular senescence is shown in Figure below. When a cell is damaged, several scenarios are possible. An antiproliferative response can be activated by which the cell becomes apoptotic or it can enter senescence. If this does not occur, the cell is able to continue replication and may form a lesion. At this stage again, both apoptotic and/or senescence programs can be activated. However, when this fails, the lesion is able to grow, cells may gain additional genetic and/or epigenetic aberrations and eventually a malignant tumor may be formed. Even if cellular senescence is induced, cells can potentially bypass the senescent state (e.g., by epigenetic changes, etc.) and undergo malignant transformation.
Schematic overview of antitumor protection by cellular senescence:
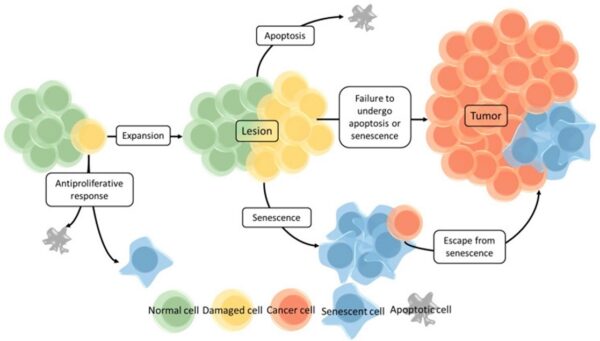
Damaged cells can become apoptotic, enter the state of senescence (antiproliferative responses) or continue to replicate (expansion). When the latter occurs, a lesion may form where cells, again, can become apoptotic or enter the state of senescence. If suitable defense mechanisms are absent or fail, the lesion can further expand and by gaining additional mutations, a cancerous lesion (tumor) may be formed. Moreover, senescent cells still can escape this state and become cancerous as well. Normal cells are indicated in green, damaged cells in yellow, cancer cells in red, senescent cells in blue, and apoptotic cells in grey.
_
Although cellular senescence is involved in normal development and ensures tissue homeostasis by limiting the growth of damaged cells, it can have detrimental effects as well. With aging, there is an accumulation of senescent cells resulting in tissue aging and eventually failure of organ homeostasis and function. Cellular senescence has been recognized as an important hallmark of aging. Senescent cells accumulate during the aging process and exhibit a senescence-associated secretory phenotype (SASP); this means that they secrete inflammatory mediators (e.g., interleukin (IL)-6, IL-8, monocyte chemoattractant protein (MCP)-2, growth-regulated oncogene alpha (GROα), etc.) that may promote tumor growth by creating a tumorigenic environment.
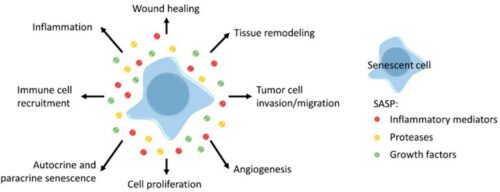
Figure above shows overview of the important SASP functions. SASP components induce wound healing, tissue remodeling and recruit immune cells. Protumor effects of the SASP are tumor cell invasion and/or migration, stimulation of vessel formation, induction of cell proliferation, and the development of an inflammatory environment. Different SASP factors establish autocrine and paracrine senescence.
_______
-3. Immunosenescence
Finally, a progressive decay of immune function occurs in older individuals, whereby an effective immune response against developing tumors may fail. A functional immune system is highly important in the prevention of tumor growth. However, aging leads to a progressive decay of immune functions, referred to as immunosenescence. Due to immunosenescence, an effective immune response against a developing tumor may fail. Moreover, it has been considered as one of the leading causes of many other age-related diseases. A strong interconnection between immunosenescence and inflammation exist, both appear to maintain and induce each other.
_
Innate Immunosenescence:
Although aging mainly alters the adaptive immune system, the innate immune system is reshaped as well (Figure below). Noteworthy, the innate immune system is activated by pathogens but also by an inflammatory response. In addition, cells of the innate immune system are able to release inflammatory cytokines and chemokines, highlighting the link with inflammation. With aging, the number of neutrophils remains relatively stable. However, neutrophil function is altered, as demonstrated by reduced chemotaxis, phagocytosis, signaling, and increased susceptibility to apoptosis. There are age-related shifts within the monocyte subpopulation, for instance the proinflammatory monocytes (CD16+) increase and the phagocytic monocytes (CD16−) decrease. This is accompanied by many functional changes of monocytes/macrophages e.g., reduced chemotaxis, phagocytosis, antigen presentation, and increased production of inflammatory cytokines. In dendritic cells, antigen presentation and signaling are reduced, and myeloid dendritic cells produce more proinflammatory cytokines. The total number of NK-cells increases with aging, and shifts in the subpopulations are seen i.e., a decrease of the immature, immunoregulatory (CD56bright) and an increase of the mature, cytotoxic NK-cells (CD56dim). As a result, response to cytokines and signaling is impaired, and the NK cells produce less cytokines and become less cytolytic. The altered functions of innate immune cells with aging may have an effect on the adaptive immune system, which is activated when an antigen, mostly presented by antigen presenting cells of the innate immune system, is recognized by T- and/or B-cells.
_
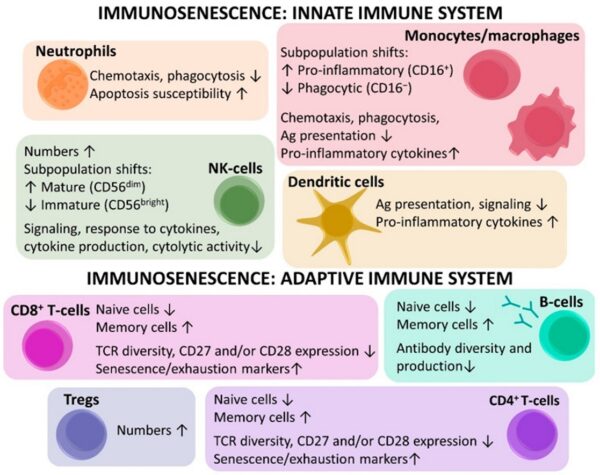
Figure above is overview of the age-related changes in the innate immune system, including neutrophils, monocytes, NK-cells, dendritic cells, and adaptive immune system with the cytotoxic CD8+ T-cells (most affected by aging), CD4+ T-helper cells, Tregs, and B-cells.
_
Adaptive Immunosenescence:
The adaptive immune system is composed of T-cells, mainly mediating cellular immunity, and B-cells, which play a major role in antibody-mediated or humoral immunity. With aging, shifts in both numbers and functions of adaptive immune cells occur (Figure above). In general, immunosenescence of the adaptive immune system is characterized by a decrease of naive cells and an accumulation of memory cells. These changes are most striking in the T-cell population, with more pronounced age-related alterations in the cytotoxic T-cells (CD8+) compared to the helper T-cells (CD4+). With aging, pluripotent hematopoietic stem cells (HSC), from which all immune cell types originate, are more likely to differentiate into myeloid than lymphoid cell lines. More importantly, however, is the thymic involution after puberty, which results in a reduced production of naive T-cells and lowered T-cell receptor (TCR) diversity. Moreover, the proliferative capacity diminishes and a lifelong exposure to numerous immune challenges further reduces TCR diversity and depletes the naive T-cell reserves by conversion into memory T-cells. Various studies have demonstrated the alterations in the T-cell compartment by evaluating different subset markers. Aging is associated with an alteration of the CD4/CD8 ratio and expansion of the immunosuppressive regulatory T-cells (Tregs). Naive, central memory (CM), effector memory (EM) and terminally differentiated effector memory (TEMRA) cells are identified by assessing the expression of CD45RA and CC-chemokine receptor 7 (CCR7). The costimulatory receptors CD27 and CD28 are important markers of T-cell activation, and CD57 and killer lectin-like receptor G1 (KLRG1) are markers of senescence. With age, Koch et al. saw a decrease in naive T-cells (CD45RA+, CCR7+) and an expansion of TEMRA T-cells (CD45RA+, CCR7−), which was more pronounced in the CD8+ as compared to the CD4+ compartment. Additionally, the expression of the costimulatory receptors CD27 and CD28 declined, especially in CD8+ T cells, and cells with lower expression of CD27 and CD28 often had a higher expression of CD57 and KLRG1. Hence, with age, numbers of naive T-cells decrease while the frequency of late-stage differentiated/senescent memory T-cells increases. Apart from an increase in the expression of senescence markers, aging can also be associated with an increase in the expression of markers of exhaustion, which include inhibitory immune checkpoints (e.g., programmed cell death protein 1 (PD-1), cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), etc.).
Although age-related changes in the B-cell population are less apparent, certain modifications could be identified as well. Like T-cells, B-cells originate from HSC, thus the age-related shift from lymphopoiesis to myelopoiesis also affects the B-cells. There is also a decrease in pre-B-cells and an increase in memory B-cells, the diversity and production of antibodies diminishes which eventually leads to altered B-cell functions.
_____
As the aging rate is unique, biological age of a person can differ from the chronological age and two people of the same calendar age can show a different biological aging profile. Consequently, one individual could tolerate more aggressive treatments than another individual of the same age could. In addition, chemotherapy by itself is also believed to accelerate the aging process, which may result in premature aging and frailty. Additionally, more extensive and intensive research is needed to gain a better understanding of the impact of aging on tumor immunity. As aging has a substantial impact on the immune system, it could influence the effectiveness of the upcoming immunotherapies. As for now, we do not have optimal tools available for estimating the “biological” age of patients, which is far more relevant than their chronological age.
______
______
Meta-hallmarks of aging and cancer, a 2023 study:
Both aging and cancer are characterized by a series of partially overlapping “hallmarks” that we subject here to a meta-analysis. Several hallmarks of aging (i.e., genomic instability, epigenetic alterations, chronic inflammation, and dysbiosis) are very similar to specific cancer hallmarks and hence constitute common “meta-hallmarks,” while other features of aging (i.e., telomere attrition and stem cell exhaustion) act likely to suppress oncogenesis and hence can be viewed as preponderantly “antagonistic hallmarks.” Disabled macroautophagy and cellular senescence are two hallmarks of aging that exert context-dependent oncosuppressive and pro-tumorigenic effects. Similarly, the equivalence or antagonism between aging-associated deregulated nutrient-sensing and cancer-relevant alterations of cellular metabolism is complex. The agonistic and antagonistic relationship between the processes that drive aging and cancer has bearings for the age-related increase and oldest age-related decrease of cancer morbidity and mortality, as well as for the therapeutic management of malignant disease in the elderly.
_____
_____
Age-related epigenetic changes on cancer development:
Age is a prominent risk factor for most types of cancer, including breast, lung and colon cancers, which each have a large impact on public health (de Magalhães, 2013). Cancer risk increases with age, in part, because genetic mutations that arise from DNA replication errors and exposure to environmental carcinogens accumulate as we get older (Tomasetti et al., 2017).
_
Aging also alters the epigenome, the chemical marks spread across DNA that help switch genes on or off by altering how the genome is packaged. For instance, the addition of a methyl group to DNA can play a role in compressing the nearby DNA sequence so it can no longer be accessed by the cell’s machinery. Epigenetic modifications, including DNA methylation, have also been shown to contribute to the development of cancer (Flavahan et al., 2017; Saghafinia et al., 2018). However, the potential impact of age-related epigenetic changes on cancer development has not been fully characterized.
_
Previous studies have identified specific DNA methylation sites that are associated with age (Horvath and Raj, 2018). Researchers have developed algorithms, called ‘epigenetic clocks’, that use data from tens to hundreds of these methylation sites to estimate an individual’s ‘epigenetic age’. This includes the Horvath clock which predicts age using DNA methylation data from any tissue type (Horvath, 2013), and the Hannum clock which was designed to use data from blood cells (Hannum et al., 2013).
_
It has been hypothesized that people whose epigenetic age is greater than their age in years – a phenomenon known as accelerated aging – may be at higher risk of age-related diseases, including cancer (Yu et al., 2020). However, previous studies linking accelerated epigenetic aging and cancer have produced mixed results. Now, in eLife, Fernanda Morales Berstein from the University of Bristol and co-workers (who are based at various institutes in the United Kingdom, the United States, Greece and Australia) report how they tackled this question using a different approach to most prior studies called Mendelian randomization (Morales Berstein et al., 2022). Instead of associating a person’s risk of cancer with epigenetic clock estimates, they correlated it against genetic variations that are known to influence these algorithms.
_
First, the team examined results from a previously conducted genome-wide association study which had analyzed the DNA of over 34,000 individuals to identify genetic variations that influence epigenetic clocks (McCartney et al., 2021). They used these results to select specific variants that predict the epigenetic age values measured by four common clocks (Horvath, Hannum, PhenoAge and GrimAge).
Next, Morales Berstein et al. used the Mendelian randomization method to find out if the variants that predict accelerated aging also affect the risk of several different types of cancer (breast, prostate, ovarian, colorectal and lung cancer). To do this the team obtained data from several large genome-wide association studies that had searched the genome of individuals for differences that predict cancer status; genetic variations related to the aging clocks were then extracted to see if they were also associated with an increased risk of cancer.
_
The results of Morales Berstein et al. did not show many clear relationships between the epigenetic aging clocks and risk for the various types of cancer studied. The most promising finding was an association between the GrimAge clock and colorectal cancer. The GrimAge clock was not designed to predict age alone, but also reflects the effects of smoking and other mortality-related epigenetic features (Lu et al., 2019). Thus, the interpretation of this association is not straightforward, as this clock may capture the effects of environmental or lifestyle factors on the epigenome.
_
While the Morales Berstein et al. study did not show pervasive effects of epigenetic aging on cancer risk, their work is a critic al contribution to cancer susceptibility research, as they have addressed an important question using rigorous methods, including Mendelian randomization. The primary strength of studies that use this approach is that they are less prone to certain types of biases that can affect observational research, such as confounding and reverse causation. Furthermore, it is important to acknowledge that epigenetic clocks have largely been developed based on how aging affects DNA methylation in blood cells. Much less is known regarding aging and epigenetics in other tissue types, including those prone to cancer, such as the ones examined by Morales Berstein et al.
_
Future studies will likely use Mendelian randomization to address similar hypotheses for additional cancer types and a wider variety of epigenetic aging algorithms. As the size of genome-wide association studies increase and more clock-related genetic variants are discovered, this approach will gain more power to detect the effects of epigenetic aging on cancer and other age-related diseases.
_____
_____
Section-6
Cancer and Tobacco (smoking and smokeless):

_
Tobacco use is the leading preventable cause of cancer and cancer deaths. It can cause not only lung cancer — but also cancers of the mouth and throat, voice box, esophagus, stomach, kidney, pancreas, liver, bladder, cervix, colon and rectum, and a type of leukemia. Tobacco smoke causes 90% of lung cancer. Tobacco smoke contains over fifty known carcinogens, including nitrosamines and polycyclic aromatic hydrocarbons. Tobacco is responsible for about one in five cancer deaths worldwide and about one in three in the developed world. Lung cancer death rates in the United States have mirrored smoking patterns, with increases in smoking followed by dramatic increases in lung cancer death rates and, more recently, decreases in smoking rates since the 1950s followed by decreases in lung cancer death rates in men since 1990. Each year, 660,000 people in the US are diagnosed with and 343,000 people die from a cancer related to tobacco use. More than 1 million tobacco-related cancer deaths have been avoided since 1990 because of comprehensive cancer and tobacco control programs, early detection of cancer, and improvements in cancer treatment. However, not all states or all people have experienced the benefits of these efforts. When states make greater and longer investments in comprehensive cancer and tobacco control programs, fewer people use tobacco and get or die from tobacco-related cancers.
Key facts:
-Tobacco use causes at least 12 types of cancer.
-Cancers linked to tobacco use make up 40% of all cancers diagnosed.
-Cigarette smoking causes 3 in 10 of all cancer deaths.
_
People who use tobacco or are exposed to second-hand smoke are more likely to get and die from cancer.
- Tobacco smoke has at least 70 chemicals that cause cancer, also known as carcinogens.
- Lung and colorectal cancers make up more than half of all cancers linked to tobacco use.
- Second-hand smoke exposure causes about 7,300 lung cancer deaths among non-smoking adults each year.
- Quitting tobacco use at any age can reduce the risk of getting or dying from cancer.
_____
_____
Table below is summary of cancers for which tobacco use (smoking and smokeless) has a definite effect on risk:
|
Relative risk for ever smoking |
Attributable risk |
Dose–response |
Cessation |
Tobacco types |
|
|
|
Lung |
15 for small cell and squamous |
85% in men and |
Intensity and duration, |
Sharp decline in risk on quitting, |
All smoking tobacco products and |
|
|
|
||||||
|
Larynx |
10–15 |
67% in men and 28% in women |
Intensity and duration |
Risk decreases after quitting to near that of never-smokers |
All forms of tobacco |
|
|
|
||||||
|
Bladder |
3 |
37% in men and 14% in women |
Intensity and duration |
Risk decreases up to 60% after quitting |
Cigarettes more deleterious than cigars or pipes and no effect of smokeless tobacco |
|
|
|
||||||
|
Oesophagus |
2–5 for squamous cell carcinoma and <2 for adenocarcinoma |
45% in men and 11% in women |
Intensity and duration |
Risk decreases after quitting, but significant increased risk persists |
Smoking tobacco, but not smokeless tobacco |
|
|
|
||||||
|
Pancreas |
2–4 |
27% in men and 11% in women |
Intensity and duration |
Risk halved after quitting |
Cigarettes and cigars |
|
|
|
||||||
|
Kidney |
2 for renal cell carcinoma and 3 for cancer of the renal pelvis |
38% in men and 4% in women |
Intensity and duration |
Up to 25% risk reduction in long-term quitters |
Cigarettes, but not cigars, pipes or smokeless tobacco |
|
|
|
||||||
|
Oral cavity, pharynx |
2–3 |
41% in men and |
Intensity and duration |
Long-term quitters have risks |
All forms of tobacco |
|
|
11% in women |
close to never-smokers |
|
||||
|
Endometrium |
0.5 |
– |
Not always a clear effect |
Long-term quitters have risks |
Cigarettes only |
|
|
Stomach |
1.5–1.6 Risk highest for adenocarcinoma and intestinal rather than diffuse variants |
13% in men and 7% in women |
Intensity and duration |
Ex smokers have a third of the excess risk of current smokers |
All forms of tobacco |
|
|
|
||||||
|
|
||||||
|
|
||||||
______
______
Mechanisms of tobacco-related carcinogenesis:
The label `cancer’ encompasses a wide variety of diseases, all of which share the common characteristic of unregulated cell growth. Carcinogenesis, or the development of cancer, is a multistage process. For cells to be free from cell growth regulation mechanisms there must be both enabling of oncogenes (genes that stimulate cell division) and switching off of tumour suppressor genes (genes that prevent cell division), so that the cells are constantly stimulated to divide but there is no control preventing the mitosis. These effects are caused by mutations. Cells with damaged DNA are usually destroyed through apoptosis; however, aberrant cells may escape normal growth control and acquired mutations may alter apoptosis, and thereby allow the development of cancer. Carcinogenesis therefore requires multiple genetic changes, as can occur within the context of long-term, repeated exposure to genotoxic products in tobacco.
_
Clearly, there is no single mechanism of tobacco-related carcinogenesis. A variety of tobacco products exist, and the methods by which they are consumed influences the release of carcinogens and therefore the link between tobacco use and cancer causation. Furthermore, the complexity of the mixture of carcinogens in tobacco smoke means that in different individuals, different carcinogens might cause different types of damage, and there is also a random component to carcinogenesis. Carcinogens must be metabolically activated to exert their deleterious effects, but this is counteracted by the ongoing detoxification of carcinogens, so that the balance between activation and detoxification determines part of the individual susceptibility to the carcinogenic effects of tobacco. It is important to elucidate these mechanisms, because an understanding of the pathways by which tobacco use causes cancer may allow the course to be blocked at some level, even in people who continue to use tobacco.
_
The most likely pathway, based on currently available data, by which tobacco use causes cancer is outlined in Figure below. Carcinogens from tobacco products can be taken in directly through inhalation or ingestion (smokeless tobacco) and also may be absorbed into the circulation. Many compounds from tobacco are converted into reactive electrophilic metabolites by oxidative (phase I) enzymes, to allow the attachment of a conjugate by inactivating (phase II) enzymes so that the substrate becomes more hydrophilic and can be excreted from the cell more easily. Unfortunately, the substrates produced in phase I have more potential to damage DNA than the precursor chemicals, that is, the carcinogens in tobacco may become metabolically activated by phase I enzymes.
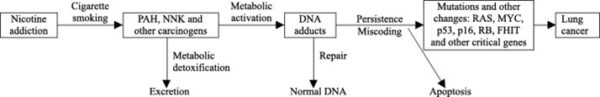
Figure above shows scheme linking nicotine addiction and lung cancer via tobacco smoke carcinogens and their induction of multiple mutations in critical genes.
Metabolites formed during the activation of carcinogens may bind covalently with DNA to form DNA adducts, usually at adenine or guanine. As evidence of this, there is a significant association between smoking status and bulky DNA adduct levels: adduct levels are highest in current smokers whilst in former smokers levels decline with years of abstinence from smoking. DNA adducts to metabolites of tobacco can also be detected in tissue not directly exposed to smoke. For instance, DNA adducts formed by benzo[a]pyrene were detected in the colonic mucosa of smokers more frequently and at higher concentrations than for nonsmokers.
If DNA adducts escape cellular repair mechanisms they could persist and may lead to miscoding, resulting in a mutation. In addition, cigarette smoke contains free radicals that can induce oxidative damage of DNA in humans and cause mutations. Certain mutations could trigger the activation of an oncogene or the deactivation of a tumour suppressor gene. The p53 gene is a key regulator of the cell cycle and mutations of the p53 gene are more common in smokers amongst lung cancer patients and oral cancer patients. These changes may allow a cell to begin dividing uncontrollably, potentially resulting in cancer causation. There is also recent evidence that nicotine might be directly implicated in carcinogenesis, beyond its crucial role in establishing and maintaining dependence, by promoting lesion growth and angiogenesis.
_
In addition to the epidemiologic evidence, experimental evidence from animals supports a role of tobacco use in carcinogenesis. Although this body of data is not without obstacles, a few examples will be given. Exposure to tobacco products, such as extracts of moist oral snuff, can produce mutations, sister chromatid exchange and chromosomal aberrations in a variety of experimental models. Furthermore, specific chemicals extracted from tobacco can be carcinogenic. For instance, some tobacco specific nitrosamines present in smokeless tobacco, such as N-nitrosonornicotine, are potent carcinogens in animal tests, producing carcinomas of the upper digestive tract and nasal cavity and the respiratory tract. Benzo[a]pyrene, which is a Polycyclic Aromatic Hydrocarbons (PAH), can also induce lung tumours upon local administration or inhalation. In addition to this, smoke condensate is clearly carcinogenic in animals. In mice and rabbits application of cigarette smoke condensate to skin induces skin cancer, and intrapulmonary injection of smoke condensate induces lung cancer in rats. Whole smoke and its particulate phase can also trigger malignant respiratory-tract tumours in hamsters and rats.
Aside from this pathway, smoking may also have an effect on the endocrine system and so influence the occurrence of hormone-related cancers. For instance, smoking may have antioestrogenic effects that could protect women from endometrial, and potentially breast cancer. Smoking may also act to reduce levels of circulating testosterone, potentially reducing the risk of prostate cancer.
_
Tobacco smoking may result in impairment of the immune system functioning thereby increasing the risk of cancer as seen in the figure below. Evidence supporting this assertion comes from both human and experimental models. Smokers have higher rates of infection, lower serum levels of most immunoglobulin classes (except for IgE) and lower antibody titres when infected. When animals are exposed to smoke they experience a suppression of their primary antibody response as well as an increased susceptibility to infections. There are a number of proposed mechanisms for this immunological effect. Cigarette smoke and/or nicotine may influence the hypothalamo-pituitary-adrenal axis by stimulating the release of catecholamines and ACTH, they may modulate cytokine production and thus change the Th1/Th2 ratio, or they could reduce the responsiveness of T cells.
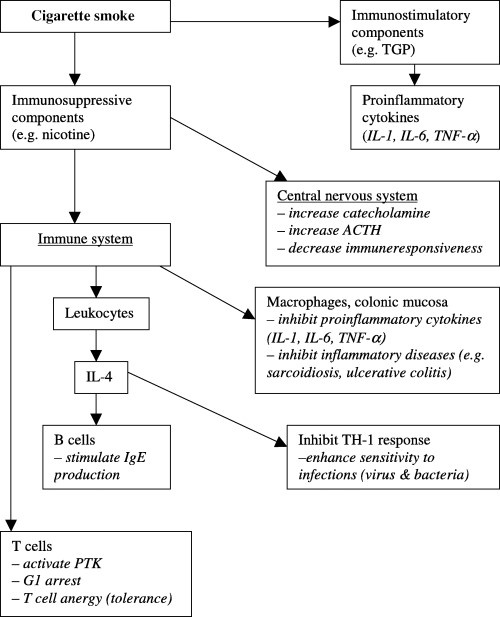
Figure above shows a simplified model by which cigarette smoke could affect the immune system.
_
The association between tobacco use and cancer varies by anatomic site. This may be the result of cofactors, such as alcohol and diet, that are present in various parts of the body, or because the duration of contact of specific anatomical sites to tobacco constituents and their metabolites varies, as well as the concentration of this contact. For instance, metabolites are excreted in the urine, and as urine is stored in the bladder substantially longer than in the kidneys this may explain the stronger effect of smoking on bladder than kidney cancer. In addition, the variation in proliferative ability of tissues may be important.
_______
Passive smoking:
Several sources of evidence suggest that passive smoking, or exposure to environmental tobacco smoke, increases the risk of cancer. Haemoglobin adducts of 4-aminobyphenyl, a carcinogen in tobacco smoke, have been identified in people exposed to environmental tobacco smoke. Moreover, side stream smoke contains more carcinogens, although at a lower concentration, than mainstream smoke and no safe limit for carcinogens exists. Epidemiologic studies have repeatedly shown an association between exposure to passive smoke and lung cancer risk in nonsmokers. The magnitude of the excess risk amongst those exposed is probably in the order of 20%. There are, of course, difficulties in inferring a causal effect of passive smoking on cancer development, as this exposure is liable to misclassification and there is likely to be confounding by other exposures related to cancer. However, the combination of biological plausibility and epidemiologic evidences makes such an association highly likely.
_______
Cofactors and tobacco-related carcinogenesis:
The fact that not all tobacco-users develop cancer is frequently used to argue against a role of tobacco in carcinogenesis. The real reason for this (aside from the random element) is that characteristics relating to individual smokers may put them at increased risk of cancer, or indeed, may be protective, and these are called cofactors. The interaction might take place at the stage of external exposure (e.g. the other agent has to be absorbed on the tobacco particles to penetrate into the lung) or at some stage of the carcinogenic process (e.g. induction of common activating or detoxifying enzymes). The potential importance of cofactors must not, however, detract from the primary role of tobacco in cancer causation.
_
Dietary, occupational and environmental cofactors:
Dietary components can modify the role of tobacco in carcinogenesis. As one example, high fruit and vegetable intake may protect from the deleterious effects of smoking with respect to lung cancer and gastric cancer. A cross-sectional study of 63 healthy male smokers revealed that blood levels of vitamin E and vitamin C and retinol, α-tocopherol and β-carotene were inversely associated with PAH–DNA adducts in circulating mononuclear cells. Hence, fruit and vegetables may protect smokers from cancer by reducing the formation of adducts, perhaps by inhibiting DNA and chromosomal damage by carcinogens and altering expression of metabolic enzymes.
_
Alcohol may confound the association between smoking and cancer, but it also acts as an effect modifier. Alcohol consumption appears to interact significantly with tobacco use in the development of oesophageal squamous-cell carcinoma, cancer of the oral cavity, pharynx and larynx, gastric cancer and liver cancer. The effect modification by alcohol may be because ethanol increases the tissue penetration or metabolic activation of tobacco smoke carcinogens. Microorganisms are important carcinogenic agents, and they may interact with smoking during carcinogenesis, as in the case of infection with Helicobacter pylori which interacts with smoking in the development of gastric cancer.
_
Occupational and environmental exposures could influence the carcinogenic effect of smoking. A less than multiplicative interaction for the effect of smoking on risk of lung cancer has been suggested in studies of workers exposed to asbestos or crystalline silica. The results of studies of uranium miners are compatible with a multiplicative interaction between smoking and radon decay products. Furthermore, nickel or arsenic appear to have an additive effect with smoking on risk for lung cancer, and smoking has a multiplicative interactive effect with occupational exposure to aromatic amines in the causation of bladder cancer.
_
Host and genetic cofactors:
Cancer is more common in older age groups. This is chiefly a function of increased duration of exposure to carcinogens, and partly because of a reduced ability to repair DNA damage with increasing age. Furthermore, risk of cancer amongst smokers may vary between men and women, either because exposure to smoking depends on gender, or because of the interactive effect of hormonal status with smoking in carcinogenesis. For instance, bladder cancer may be more strongly related to smoking amongst women than men and risk of smoking is particularly protective for endometrial cancer amongst women with high levels of oestrogen.
There are also established racial differences in tobacco-related diseases; for example, there may be a closer association between bladder cancer risk and smoking in African Americans than White Americans. Experimental evidence has shown that nicotine intake per cigarette is 30% greater in Black than White smokers, moreover Black smokers clear cotinine at a significantly slower rate, possibly because of a poorer potential for detoxification.
_
Hereditary variation in the metabolic pathways through which carcinogens are activated and detoxified appear to influence risk for developing cancer. A good model of this is provided by the Cytochrome P450 (CYP) proteins that comprise many of the phase I enzymes. A number of CYP proteins exist, and polymorphisms in these proteins may relate to cancer risk amongst tobacco users. For example, CYP1A1 probably plays an important role in the activation of PAHs. Certain CYP1A1 variants are more efficient at bioactivating procarcinogens, and these variants confer a higher susceptibility to lung cancer.
Furthermore, CYP2A6 activates NNK and certain N-nitrosamines. This CYP2A6 activity varies considerably in humans, partly because variant CYP2A6 alleles code for the inactive enzyme. The frequency of CYP2A6 whole deletion was found to be lower in nonsmall cell lung cancer cases than controls. There is, therefore, evidence to suggest that diminished CYP activity might decrease the production of carcinogenic metabolites, and thereby reduce the risk of cancer amongst smokers.
_
Near complete loss of N-acetyltransferase 2 (NAT2) enzyme activity is caused by at least seven different alleles resulting from single-base substitution. People with two mutant alleles are slow acetylators, with a reduced ability to detoxify aromatic amines, whereas those with at least one normal allele are fast acetylators. Studies have shown that smoking is significantly more deleterious, with respect to risk to bladder cancer, and possibly colorectal cancer, amongst people with the NAT2 slow acetylation polymorphism.
The glutathione S-transferase μ gene (GSTM1) is another genetic susceptibility factor that codes for a phase II detoxification enzyme. Within the group of smokers, PAH–DNA adducts in circulating mononuclear cells were detected less frequently amongst those with an inherited absence of GSTM1 (the null GSTM1 genotype). Indeed, studies have shown that people with the null GSTM1 genotype may have a significantly higher risk of, inter alia, lung and laryngeal cancer, probably on account of their reduced ability to detoxify carcinogens.
_
A number of genes have been identified that confer highly increased risk for specific types of cancer, and these are known as high penetrance cancer genes. These high penetrance cancer genes could alter the effect of tobacco use in promoting carcinogenesis. For instance, two highly penetrant genes for breast cancer have been identified, the breast cancer 1 (BRCA1) and 2 (BRCA2) genes, which are associated with a very high risk of breast cancer. In a matched case–control study of female carriers of either BRCA1 or BRCA2 mutation, women with breast cancer were half as likely to have smoked cigarettes at any time in their lives as healthy control subjects, and a dose–response protective effect of smoking duration and intensity was observed. This protective effect may be the result of the antioestrogenic effect of smoking.
_____
_____
Section-7
Diet and cancer:

_
The field of investigation of the role of nutrition in the cancer process is very broad. It is becoming clearer as research continues that nutrition plays a major role in cancer. It has been estimated by the American Institute for Cancer Research and the World Cancer Research Fund that 30–40 percent of all cancers can be prevented by appropriate diets, physical activity, and maintenance of appropriate body weight. It is likely to be higher than this for some individual cancers.
Most of the research on nutrition and cancer has been reductionist; that is, a particular food or a nutrient has been studied in relation to its impact on tumor formation/regression or some other end point of cancer at a particular site in the body. These studies are very helpful in seeing the details of the mechanisms of disease. However, they do not help give an overall picture of how to prevent cancer on a dietary level. Even less, they tell little of how to eat when a person already has a cancer and would like to eat a diet that is favorable to their recovery.
_
While many dietary recommendations have been proposed to reduce the risk of cancer, few have significant supporting scientific evidence. Obesity and drinking alcohol have been correlated with the incidence and progression of some cancers. Lowering the drinking of beverages sweetened with sugar is recommended as a measure to address obesity. A diet low in fruits and vegetables and high in red meat has been implicated but not confirmed, and the effect may be small for well-nourished people who maintain a healthy weight. Some specific foods are linked to specific cancers. Studies have linked eating red or processed meat to an increased risk of breast cancer, colon cancer, prostate cancer, and pancreatic cancer, which may be partially explained by the presence of carcinogens in foods cooked at high temperatures. Aflatoxin B1, a frequent food contaminant, increases risk of liver cancer, while drinking coffee is associated with a reduced risk. Betel nut chewing causes oral cancer. Stomach cancer is more common in Japan due to its high-salt diet. Immigrant communities tend to develop the risk of their new country, often within one generation, suggesting a substantial link between diet and cancer. Dietary recommendations for cancer prevention typically include weight management and eating “mainly vegetables, fruit, whole grains and fish, and a reduced intake of red meat, animal fat, and refined sugar.” Large claims have been made for the effectiveness of particular diets in preventing cancer or inhibiting its progression. However, more recent clinical studies have not confirmed this. Instead, it seems that rather than specific dietary constituents, total calories influence cancer incidence and progression.
______
Molecular mechanisms:
For a long time, somatic mutagenesis stood at the centre of interest in the field of carcinogenesis. As a consequence, mutagenic dietary factors and nutrients that impair gene repair were investigated in detail. Among those identified are natural compounds consumed directly (e.g., in herbal teas) or indirectly via herbivores feeding on bracken fern for example. Food might also be contaminated with mutagenic molecules, with the best known example being aflatoxin produced by Aspergillus flavus growing on peanuts and cereals. Finally, heterocyclic amines, polycyclic aromatic hydrocarbons, N-nitroso-compounds or acrylamide can be formed by the process of cooking, particularly of meat. The picture is complicated, however, by dietary factors that oppose the action of mutagens. Figure below shows that mTOR and AMPK oppose each other.
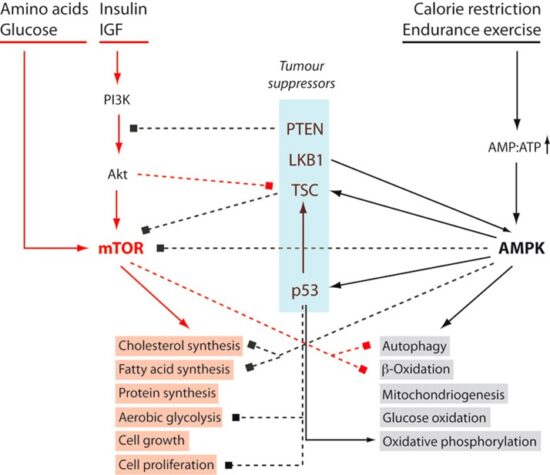
Tumour enhancing pathways are coloured red, inhibitory interactions are represented by dashed lines.
AMPK: AMP-dependent kinase; IGF: insulin-like growth factor; LKB1: Serine/threonine-protein kinase 11; mTOR: mammalian target of rapamycin; PI3K: phosphoinositide 3-kinase; PTEN: phosphatase and tensin homologue; TSC: tuberous sclerosis complex.
In figure above, two opposing hubs of the metabolic signalling network are placed at the centre of events: the “mammalian Target of Rapamycin” (mTOR), a serine protein kinase which stimulates anabolic, ATP-consuming processes such as protein synthesis, lipid synthesis and cell growth (the “red block” in figure above); and the adenosine monophosphate (AMP)-dependent kinase (AMPK) which stimulates catabolic, ATP-generating processes, particularly in the mitochondrion (the “grey block” in figure above). Both are “energy sensors” with mTOR responding to high levels of energy carriers, glucose and amino acids, as well as to growth factors (insulin, insulin-like growth factor 1 etc.), and with AMPK responding to an increase of the AMP:ATP ratio (i.e. a slight depletion of the intracellular energy source ATP). The two pathways are not only activated by opposite metabolic situations, but they also inhibit each other. AMPK is thought to be a major element or mechanism in cancer-related effects of diet. It modulates the activity of cellular survival signaling such as mTOR and Akt, leading to cell growth inhibition which is relevant to cancer growth.
It is clear that merely shifting the balance in favour of mTOR is not enough to cause cancer. However, it should be noted that four of the signalling intermediates are tumour suppressors that, directly or indirectly, inhibit mTOR: PTEN (phosphatase and tensin homologue), LKB1 (serine/threonine-protein kinase 11), TSC (tuberous sclerosis complex) and p53. It might, therefore, be expected that, in the long run, excessive mTOR activity will increase the probability of carcinogenesis.
High fasting blood glucose, insulin and insulin-like growth factor (IGF) concentrations stimulate the mTOR pathway and are also features of metabolic syndrome. The latter is known to positively correlate with cancer incidence, and so a diet preventing obesity and metabolic syndrome reduces the likelihood of developing cancer. Although the role of mTOR pathways in carcinogenesis is emphasized, this is the not the only mechanism explaining increased cancer incidence in obesity and the metabolic syndrome. There is, for example, abundant evidence that inflammatory adipokines released by visceral adipose tissue may favour tumorigenesis.
_
Methionine metabolism and cancer:
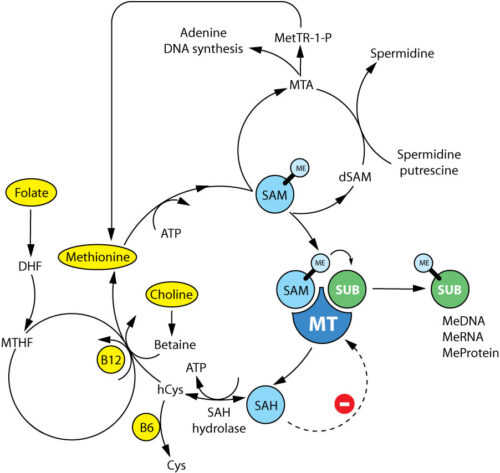
Figure above shows methionine metabolism pathway.
DHF, dihydrofolate; dSAM, decarboxylated S-adenosylmethionine; hCys, homocysteine; ME, methyl group; MetTR-1-P, 5-methylthioribose-1-phosphate; MT, methyltransferase; MTA, methylthioadenosine; MTHF, methylenetetrahydrofolate; SAH, S-adenosyl-L-homocysteine; SAM, S-adenosyl methionine; SUB, substrate.
Although numerous cellular mechanisms are involved in food intake, many investigations over the past decades have pointed out defects in the methionine metabolic pathway as cause of carcinogenesis. For instance, deficiencies of the main dietary sources of methyl donors, methionine and choline, lead to the formation of liver cancer in rodents. Methionine is an essential amino acid that must be provided by dietary intake of proteins or methyl donors (choline and betaine found in beef, eggs and some vegetables). Assimilated methionine is transformed in S-adenosyl methionine (SAM) which is a key metabolite for polyamine synthesis, e.g. spermidine, and cysteine formation (see the figure on the right). Methionine breakdown products are also recycled back into methionine by homocysteine remethylation and methylthioadenosine (MTA) conversion (see the figure on the right). Vitamins B6, B12, folic acid and choline are essential cofactors for these reactions. SAM is the substrate for methylation reactions catalyzed by DNA, RNA and protein methyltransferases.
The products of these reactions are methylated DNA, RNA or proteins and S-adenosylhomocysteine (SAH). SAH has negative feedback on its own production as an inhibitor of methyltransferase enzymes. Therefore, SAM:SAH ratio directly regulates cellular methylation, whereas levels of vitamins B6, B12, folic acid and choline regulates indirectly the methylation state via the methionine metabolism cycle. A near ubiquitous feature of cancer is a maladaption of the methionine metabolic pathway in response to genetic or environmental conditions resulting in depletion of SAM and/or SAM-dependent methylation. Whether it is deficiency in enzymes such as methylthioadenosine phosphorylase, methionine-dependency of cancer cells, high levels of polyamine synthesis in cancer, or induction of cancer through a diet deprived of extrinsic methyl donors or enhanced in methylation inhibitors, tumor formation is strongly correlated with a decrease in levels of SAM in mice, rats and humans.
According to a 2012 review, the effect of methionine restriction on cancer has yet to be studied directly in humans and “there is still insufficient knowledge to give reliable nutritional advice”.
_______
Methodological aspects:
Establishing a causal relationship between a particular diet and cancer is difficult. Cancer is a chronic disease surfacing after many years of (possibly) harmful eating behaviour. This practically excludes the kind of controlled experiments demanded in science and leaves us with mainly observational studies.
Observational studies come in two flavours: In case-control studies, two groups are compared – one suffering from a disease (the “case”) and a matched control group. Eating habits are recorded with the help of questionnaires, but obviously suffer from recall bias, difficulties in remembering past behaviour and changes in that behaviour with time. Cohort studies are prospective. The diet of disease-free individuals is recorded, and their fate is followed over the years.
Both types of observational studies are flawed. Besides the fact that a correlation does at best suggest, but never prove a causal relationship, the groups are self-chosen and therefore most likely hide numerous additional differences in diet and lifestyle such as health-consciousness, physical activity and smoking. Those eating large amounts of red meat might represent the quintessential “steak and potato guys”. If a correlation is found, what causes it? Is it the red meat? Or the potatoes? Or maybe the combination of the two? Would a hypothetical group eating red meat and cabbage also be at risk? To a certain extent, these factors can be corrected for, but only when the observer is aware of their existence.
Often, case-control studies come up with a significant correlation between a given macro- or micronutrient and cancer incidence, but cohort studies fail to confirm the result. The reason for this is not clear, but the difficulty of recalling past eating behaviour may play a role.
Not properly appreciated is the fact that many epidemiological studies which come up with a “significant” (p <0.05) relationship are wrong. In a paper provocatively entitled “Why Most Published Research Findings Are False”, Ioannidis explains why this is so. The likelihood of false positive results increases not only with small study sizes, small effects (many nutritional studies operate with relative risks of 1.1–1.5) and the prejudices of the investigators, but also with the number of possible correlational combinations typical for hypothesis-generating experiments.
Interventional studies (randomised controlled trials = RCTs) are more reliable because individuals are randomly assigned to treatment groups, excluding hidden biases introduced by self-choice. However, participants have to stick to their assigned diet which limits the time-span of the trials.
It is a very old and very common idea that by eating healthy food, cancer can be kept at bay. Many scientific and popular guidelines have been published that offer advice on what to eat, and worldwide, nutritional supplements worth 27 billion dollars are being sold every year. This stands in sharp contrast to the paucity of evidence, most of which is derived from epidemiological data. Controlled trials, on the other hand, are mostly missing.
______
The evidence for pro- and anti-cancerogenic effects of different kinds of food:
_
-1. Fat:
A diet high in fat has been suspected to contribute to carcinogenesis and a poor outcome in cancer patients since the late 1950s. Lipids and lipid metabolites play a role in oncogenic signalling, and two preclinical examples are: (i) oxidised low density lipoprotein (LDL) promotes cellular transformation through NFκB signalling; (ii) overexpression of monoacylglycerol lipase does increase migration, invasion and survival of cancer cells. However, lipids and lipid metabolism in the body don’t directly reflect lipids in the diet – they are the result of a complex interplay between all dietary components, other lifestyle factors and genetics. Whether eating a high fat diet increases cancer incidence and mortality or not needs to be established by large, adequately powered, randomised and controlled interventional trials. No such study has yet been performed. Nonetheless, case-control and cohort studies have shed some light on the question.
Colorectal cancer:
Studies investigating the effect of total fat intake on colon cancer incidence have been inconsistent. Usually, case-control studies have been more likely to find a positive correlation than cohort studies. The Women’s Health Initiative randomised controlled trial found no such association. In most observational studies, the association between total fat intake and colorectal cancer incidence disappeared after an adjustment for total calories. This may be due to the strong correlation between calorie and total fat intake in industrialised countries. Interestingly, one cohort study even found a reduced incidence of colorectal cancer in women with a high consumption of high-fat dairy food. The benefit might be mediated by conjugated linoleic acids, but there are many other compounds in milk whose influence on carcinogenesis is unaccounted for. In addition, there are some limited data that consumption of fish and w-3 long chain polyunsaturated fatty acids may be beneficial.
Prostate cancer:
Several case-control and cohort studies have not been able to establish a link between total fat uptake and prostate cancer risk. There are not enough data to draw a conclusion regarding the effect of specific classes of fatty acids on prostate cancer incidence.
Breast cancer:
The Women’s Health Initiative study showed a risk reduction with a low fat diet, which was borderline significant. However, other case-control and cohort studies have produced conflicting results. Again, it is total energy rather than total fat uptake that has an impact on the frequency of breast cancer.
Prompted by animal data, many case-control and cohort studies investigating the effect of specific fatty acids on breast cancer development have been performed. The results of these studies are prone to confounding effects due to the different possible sources of fatty acids. Based on the current data, no firm conclusion can be drawn regarding the contribution of specific fatty acids to breast cancer incidence.
Taken together, in as much as fat uptake contributes to obesity, it may be correlated with a higher cancer risk. However, there is no convincing evidence that fat uptake per se (i.e. independent from total energy uptake and obesity) is a risk factor for malignant disease.
______
-2. Meat:
Red meat refers to all types of mammalian muscle meat, such as beef, veal, pork, lamb, mutton, horse, and goat. Processed meat refers to meat that has been transformed through salting, curing, fermentation, smoking, or other processes to enhance flavour or improve preservation. Most processed meats contain pork or beef, but processed meats may also contain other red meats, poultry, offal, or meat by-products such as blood. Examples of processed meat include hot dogs (frankfurters), ham, sausages, corned beef, and biltong or beef jerky as well as canned meat and meat-based preparations and sauces. Meat (especially when red or processed) is counted among the “usual suspects” contributing to carcinogenesis.
_
The International Agency for Research on Cancer (IARC), the cancer agency of the World Health Organization, has evaluated the carcinogenicity of the consumption of red meat and processed meat in 2015.
Red meat:
After thoroughly reviewing the accumulated scientific literature, a Working Group of 22 experts from 10 countries convened by the IARC Monographs Programme classified the consumption of red meat as probably carcinogenic to humans (Group 2A), based on limited evidence that the consumption of red meat causes cancer in humans and strong mechanistic evidence supporting a carcinogenic effect. This association was observed mainly for colorectal cancer, but associations were also seen for pancreatic cancer and prostate cancer.
Processed meat:
Processed meat was classified as carcinogenic to humans (Group 1), based on sufficient evidence in humans that the consumption of processed meat causes colorectal cancer. The experts concluded that each 50 gram portion of processed meat eaten daily increases the risk of colorectal cancer by 18%.
_
Heterocyclic amines (HCAs) and polycyclic aromatic hydrocarbons (PAHs) are chemicals formed when muscle meat, including beef, pork, fish, or poultry, is cooked using high-temperature methods, such as pan frying or grilling directly over an open flame. In laboratory experiments, HCAs and PAHs have been found to be mutagenic—that is, they cause changes in DNA that may increase the risk of cancer.
HCAs are formed when amino acids (the building blocks of proteins), sugars, and creatine or creatinine (substances found in muscle) react at high temperatures. PAHs are formed when fat and juices from meat grilled directly over a heated surface or open fire drip onto the surface or fire, causing flames and smoke. The smoke contains PAHs that then adhere to the surface of the meat. PAHs can also be formed during other food preparation processes, such as smoking of meats.
HCAs are not found in significant amounts in foods other than meat cooked at high temperatures. PAHs can be found in other smoked foods, as well as in cigarette smoke and car exhaust fumes.
The formation of HCAs and PAHs varies by meat type, cooking method, and “doneness” level (rare, medium, or well done). Whatever the type of meat, however, meats cooked at high temperatures, especially above 300 ºF (as in grilling or pan frying), or that are cooked for a long time tend to form more HCAs. For example, well-done, grilled, or barbecued chicken and steak all have high concentrations of HCAs. Cooking methods that expose meat to smoke contribute to PAH formation.
HCAs and PAHs become capable of damaging DNA only after they are metabolized by specific enzymes in the body, a process called “bioactivation.” Studies have found that the activity of these enzymes, which can differ among people, may be relevant to the cancer risks associated with exposure to these compounds.
Studies have shown that exposure to HCAs and PAHs can cause cancer in animal models. In many experiments, rodents fed a diet supplemented with HCAs developed tumors of the breast, colon, liver, skin, lung, prostate, and other organs. Rodents fed PAHs also developed cancers, including leukemia and tumors of the gastrointestinal tract and lungs. However, the doses of HCAs and PAHs used in these studies were very high—equivalent to thousands of times the doses that a person would consume in a normal diet.
Population studies have not established a definitive link between HCA and PAH exposure from cooked meats and cancer in humans. One difficulty with conducting such studies is that it can be difficult to determine the exact level of HCA and/or PAH exposure a person gets from cooked meats. Although dietary questionnaires can provide good estimates, they may not capture all the detail about cooking techniques that is necessary to determine HCA and PAH exposure levels. In addition, individual variation in the activity of enzymes that metabolize HCAs and PAHs may result in exposure differences, even among people who ingest (take in) the same amount of these compounds. Also, people may have been exposed to PAHs from other environmental sources, not just food.
Numerous epidemiologic studies have used detailed questionnaires to examine participants’ meat consumption and cooking methods. Researchers found that high consumption of well-done, fried, or barbecued meats was associated with increased risks of colorectal, pancreatic, and prostate cancer. However, other studies have found no association with risks of colorectal or prostate cancer.
In 2015, an independent panel of experts convened by the International Agency for Research on Cancer (IARC) determined consumption of red meat to be “probably carcinogenic to humans” (Group 2A), based largely on data from the epidemiologic studies and on the strong evidence from mechanistic studies. However, IARC did not conclude that HCAs and PAHs were associated with cancer incidence.
_______
-3. Fruits, vegetables and dietary fibre:
One of the most important messages of modern nutrition research is that a diet rich in fruits and vegetables protects against cancer. (The greatest message is that this same diet protects against almost all other diseases, too, including cardiovascular disease and diabetes.) There are many mechanisms by which fruits and vegetables are protective, and an enormous body of research supports the recommendation for people to eat more fruits and vegetables.
_
Block et al reviewed about 200 studies of cancer and fruit and vegetable intake. A statistically significant protective effect of fruits and vegetables was found in 128 of 156 studies that gave relative risks. For most cancers, people in the lower quartile (1/4 of the population) who ate the least amount of fruits and vegetables had about twice the risk of cancer compared to those who in the upper quartile who ate the most fruits and vegetables. Even in lung cancer, after accounting for smoking, increasing fruits and vegetables reduces lung cancer; an additional 20 to 33 percent reduction in lung cancers is estimated.
_
Steinmetz and Potter reviewed the relationship between fruits, vegetables, and cancer in 206 human epidemiologic studies and 22 animal studies. They found “the evidence for a protective effect of greater vegetable and fruit consumption is consistent for cancers of the stomach, esophagus, lung, oral cavity and pharynx, endometrium, pancreas, and colon.” Vegetables, and particularly raw vegetables, were found to be protective; 85% of the studies that queried raw vegetable consumption found a protective effect. Allium vegetables, carrots, green vegetables, cruciferous vegetables, and tomatoes also had a fairly consistent protective effect. Allium vegetables (garlic, onion, leeks, and scallions) are particularly potent and have separately been found to be protective for stomach and colorectal cancers and prostate cancer. There are many substances that are protective in fruits and vegetables, so that the entire effect is not very likely to be due to any single nutrient or phytochemical. Steinmetz and Potter list possible protective elements: dithiolthiones, isothiocyanates, indole-32-carbinol, allium compounds, isoflavones, protease inhibitors, saponins, phytosterols, inositol hexaphosphate, vitamin C, D-limonene, lutein, folic acid, beta carotene (and other carotenoids), lycopene, selenium, vitamin E, flavonoids, and dietary fiber.
_
A joint report by the World Cancer Research Fund and the American Institute for Cancer Research found convincing evidence that a high fruit and vegetable diet would reduce cancers of the mouth and pharynx, esophagus, lung, stomach, and colon and rectum; evidence of probable risk reduction was found for cancers of the larynx, pancreas, breast, and bladder.
_
Many of the recent reports from prospective population-based studies of diet and cancer have not found the same protective effects of fruits and vegetables that were reported earlier in the epidemiological and case-control studies. One explanation is that people’s memory of what they ate in a case-cohort study may have been tainted by their disease state. Another problem might be that the food frequency questionnaires (FFQ) used to measure food intake might not be accurate enough to detect differences. Such a problem was noted in the EPIC study at the Norfolk, UK site. Using a food diary the researchers found a significant correlation between saturated fat intake and breast cancer, but using a FFQ there was no significant correlation. So, inaccurate measurement of fruit and vegetable intake might be part of the explanation as well.
_
Cruciferous Vegetables:
Cruciferous vegetables (broccoli, cauliflower, cabbage, Brussels sprouts) contain sulforaphane (an isothiocyanate), which has anti-cancer properties. A case-control study in China found that intake of cruciferous vegetables, measured by urinary secretion of isothiocyanates, was inversely related to the risk of breast cancer; the quartile with the highest intake only had 50% of the risk of the lowest intake group. In the Nurses’ Health Study a high intake of cruciferous vegetables (5 or more servings/week vs less than two servings/week) was associated with a 33% lower risk of non-Hodgkin’s lymphoma. In the Health Professionals Follow-up Study bladder cancer was only weakly associated with low intake of fruits and vegetables, but high intake (5 or more servings/week vs 1 or less servings/wk) of cruciferous vegetables was associated with a statistically significant 51% decrease in bladder cancer. Also, prostate cancer risk was found to be reduced by cruciferous vegetable consumption in a population-based case-control study carried out in western Washington state. Three or more servings per week, compared to less than one serving of cruciferous vegetables per week resulted in a statistically significant 41% decrease in prostate cancer risk. Similar protective effects of cruciferous vegetables were seen in a multi-ethnic case-control study. A prospective study in Shanghai, China found that men with detectable amounts of isothiocyanates in their urine (metabolic products that come from cruciferous vegetables) had a 35% decreased risk of lung cancer. Among men that had one or two genetic polymorphisms that caused them to eliminate these isothiocyanates slower there was a 64% or 72% decreased risk of lung cancer, respectively.
Broccoli sprouts have a very high concentration of sulforaphane since this compound originates in the seed and is not made in the plant as it grows. One sprout contains all of the sulforaphane that is present in a full-grown broccoli plant. So, if sulforaphane is especially cancer-protective, it would seem reasonable to include some broccoli sprouts in an anti-cancer diet.
_
Many case-control and cohort studies are dealing with the effect of fruits and vegetables on cancer incidence. Early data indicated a beneficial effect (summarised by Block et al. vide supra) and, as recently as 2008, Freedman et al. found a reduced occurrence of head and neck cancers with increased fruit and vegetable consumption. On the other hand, a Cochrane study came up empty when correlating the amount of dietary fibre with the occurrence of colorectal cancer. Similarly, Food and Drug Administration (FDA) review found little evidence to support an association between tomato consumption and cancer risk. Lifestyle issues are powerful confounding factors when investigating the effect of fruits, vegetables and dietary fibre on health. Even after carefully adjusting for these factors, residual uncertainties remain.
Four large prospective trials have assessed the correlation between fruit and vegetable intake and overall cancer risk. In a European study, after 8.7 years of follow-up a higher intake of 200 g of vegetable and fruit resulted in a slightly reduced cancer risk (hazard ratio = 0.97,). The result was significant, but not when subjected to a more stringent Bayesian analysis. These results are in line with two earlier cohort trials that reported no correlation between fruit and vegetable uptake and cancer risk. In the NIH-AARP study, fruits or vegetables were not associated with cancer incidence, except for a small inverse association between vegetable consumption and cancer in men.
The inverse correlation between the uptake of fruits and vegetables and the consumption of alcohol and tobacco is a potent confounding factor. At this point, it has to be concluded that fruits, vegetables and dietary fibre have a very marginal, if any, effect on cancer incidence.
____
-4. Vitamins, antioxidants and other micronutrients:
Many trials have investigated the correlation between the consumption of vitamins and antioxidants and the incidence of cancer. In contrast to most other studies on diet and cancer, which rely heavily on a cohort or case-control design, some randomised controlled trials are available in the case of vitamins and antioxidants. In the Women’s Antioxidants Cardiovascular Trial, vitamin C, E and β-carotene were supplemented. After a median follow up of 9.4 years, no difference in cancer incidence was noted. At the level of individual cancer types, a Cochrane review found no decrease in gastrointestinal cancers when vitamins and antioxidants were added to the diet. In addition, multivitamins were not effective in preventing recurrence or increasing overall survival in colorectal cancer patients. β-carotene did not prevent cancer, and on the contrary, it increased the risk of lung and gastric cancer in smokers and in asbestos workers (systematic review by Druesne-Pecollo).
Based on a number of case-control and cohort studies, it was claimed that supplementing the diet with folic acid or natural folates may reduce the risk of colorectal cancer. However, an effect was only seen in persons who took supplements for at least 15 years, whereas the number of polyps was not reduced. Heavy smokers and people who consume large quantities of alcohol, conditions known to deplete folate reserves, might benefit from folate supplements. In contrast, circulating folate was positively associated with the risk of prostate cancer in a meta-analysis of prospective studies showing an odds ratio of 1.18 (95% confidence interval 1.0–1.4 per 10 nmol/L increase). This suggests that there must be an ideal range of folate in the body, below and above which the cancer risk increases.
Selenium is another micronutrient studied for its cancer preventing potential. However, the SELECT trial has provided no evidence that selenium prevents cancer in the selenium replete population.
While in general, supplementation with vitamins, antioxidants or other micronutrients does neither reduce the risk of carcinogenesis nor improve the outcome in patients suffering from cancer, there is one possible exception: vitamin D was found to reduce the risk of colorectal cancer. In an unplanned subgroup analysis of the Women’s Health Initative, supplementation with calcium and vitamin D reduced the risk of melanoma in women with previous non-melanoma skin cancer. However, with regard to breast cancer, prospective cohort trials showed a much smaller benefit than retrospective case-control studies. In addition, most of the laboratory work which forms the rationale for studies on vitamin D was done with 1,25-dihydroxy-cholecalciferol, whereas in nutritional studies 25-hydroxy-cholecalciferol was supplemented. This complicates the interpretation of the results.
In summary, supplementation with vitamins, antioxidants or other micronutrients offers no relevant benefit in the primary prevention of cancer incidence and mortality. The potential benefits of vitamin D and folate have to be confirmed or disproved by additional studies, preferentially with interventional trials. Researchers from the German Cancer Research Center say daily intake of vitamin D could help reduce cancer death risk by 12%.
_____
-5. Alcohol:
Alcohol is an example of a chemical carcinogen. The World Health Organization has classified alcohol as a Group 1 carcinogen. In Western Europe 10% of cancers in males and 3% of cancers in females are attributed to alcohol. Worldwide, 3.6% of all cancer cases and 3.5% of cancer deaths are attributable to alcohol. In particular, alcohol use has been shown to increase the risk of developing cancers of the mouth, esophagus, pharynx, larynx, stomach, liver, ovaries, and colon. The main mechanism of cancer development involves increased exposure to acetaldehyde, a carcinogen and breakdown product of ethanol. Acetaldehyde induces DNA interstrand crosslinks, a form of DNA damage. These can be repaired by an inaccurate replication-coupled DNA repair pathway. This repair pathway results in increased mutation frequency and altered mutational spectrum. Other mechanisms have been proposed, including alcohol-related nutritional deficiencies, changes in DNA methylation, and induction of oxidative stress in tissues.
_
Ethanol in alcoholic beverages is carcinogenic to humans (International Agency for Research on Cancer, 2010), and the occurrence of malignant tumours of the oral cavity, pharynx, larynx, oesophagus, liver, colon, rectum and female breast is causally related to alcohol consumption (Baan et al., 2007). Many cohort and case-control studies have consistently provided evidence of the association between these cancers and alcohol consumption (World Cancer Research Fund/American Institute for Cancer Research, 2007). Although seldom fully appreciated, the relationship between alcohol consumption and cancer risk is firmly established. Alcohol is the most frequent cause of hepatocellular carcinoma (HCC), accounting for about 40% of all cases. Regular consumption of more than 80 grams of alcohol per day for more than 10 years increases the risk for HCC approximately 5-fold, approaching an absolute risk of about 1% per year in alcoholic liver cirrhosis. When adjusted for tobacco use and other potential confounding factors, alcohol consumption of more than 60 grams per day increases the relative risk for oral or hypopharyngeal squamous cell cancer 3.2 to 9.2 fold (reviewed by Goldstein et al.). Drinking moderate quantities of alcohol (up to 12.5 g/day) results in a relative risk of 1.4 for esophageal cancer. The relative risk is 2.6 for intermediate-level (>12.5 to 50 g/day) and 5.5 for high-level (>50 g per day) alcohol consumption. In colorectal cancer, there is a strong correlation with alcohol consumption as well. The relative risk is 1.21 (95% confidence interval 1.13–1.28) for moderate and 1.52 (95% confidence interval 1.27–1.81) for heavy (≥4 drinks per day) drinking. Alcohol consumption also significantly increases the risk for breast cancer. The association is stronger for the hormone-sensitive form of breast cancer than for the insensitive form. Consuming 3 or more alcoholic drinks per week also augments the risk of recurrence and cancer-related death upon diagnosis of breast cancer, but these results still await confirmation in other large, prospective studies of breast cancer survivors with long-term follow-ups. There also seems to be a modest increase in the incidence of prostate cancer if 7 or more drinks per day are consumed.
Although the studies mentioned are case-control studies – for obvious reasons, controlled randomised trials are not feasible – the very high-risk values and the consistency of the results allow us to conclude that confounding factors introduced by self-selection will not modify the interpretation substantially. Thus, ethanol and its metabolite acetaldehyde are carcinogens.
_
Long-Term Alcohol Consumption and Breast, Upper Aero-Digestive Tract and Colorectal Cancer Risk: A Systematic Review and Meta-Analysis, 2015:
Cancers of female breast, upper aero-digestive tract (UADT) (oral cavity, pharynx, larynx, oesophagus) and colorectum are causally related to alcohol consumption. Although alcohol consumption is likely to vary during life, the few studies that have explicitly measured lifetime consumption or intake over time have not been summarised. Authors therefore conducted a systematic review and meta-analysis. Sixteen articles for breast, 16 for UADT and 7 for colorectal cancer met the eligibility criteria. There was wide variation in terms of study design, the age of study participants, the methods used to capture exposure to alcohol, the units of alcohol measurement and intake categories used and the covariates adjusted for in multivariate models. Nevertheless, authors observed an increased cancer risk associated with a higher intake of alcohol over time or during lifetime: a 28% higher risk of breast cancer, approximately a 3-fold higher risk of UADT cancer, a 5-fold higher risk of cancer of oral cavity and pharynx, a 2-fold higher risk of cancer of larynx, a 7-fold higher risk of oesophageal cancer and a 49% higher risk of colorectal cancer, for the highest intake category compared with the lowest. In conclusion, authors findings reinforce an association between alcohol intake during lifetime and breast, UADT and colorectal cancer, but show that measuring lifetime intake may not substantially increase the strength of the associations between alcohol and cancers of the breast, UADT and colorectum.
The biological mechanisms whereby alcohol causes cancer are not clearly understood. Alcohol is thought to exert its action through reactive metabolites, oxidative stress followed by lipid peroxidation, epigenetic alterations due to a reduced methyl transfer, decreased retinoic acid concentrations and by interfering with oestrogen metabolism that may influence hormone levels and oestrogen receptors (World Cancer Research Fund/American Institute for Cancer Research, 2007; Seitz et al., 2012). A direct carcinogenic effect of acetaldehyde on mucosal cells (World Cancer Research Fund/American Institute for Cancer Research, 2007; International Agency for Research on Cancer, 2010) and alcohol acting as a solvent to enable the penetration of other carcinogenic molecules (Schottenfeld and Fraumeni, 2006) has also been mentioned. Acetaldehyde also increases the rate of cellular proliferation in the rectal mucosa (Seitz and Simanowski, 1988). In addition, heterozygous carriers of variant allele ALDH2*2 (which encodes an inactive subunit of an enzyme that detoxifies acetaldehyde to acetate) are at a higher risk of developing alcohol-related aero-digestive tract cancers due to higher levels of acetaldehyde (Lewis and Smith, 2005; Matsuda et al., 2006).
______
-6. Tea and coffee:
Tea is composed of polyphenols, alkaloids (caffeine, theophylline, and theobromine), amino acids, carbohydrates, proteins, chlorophyll, volatile organic compounds (chemicals that readily produce vapors and contribute to the odor of tea), fluoride, aluminum, minerals, and trace elements. The polyphenols, a large group of plant chemicals that includes the catechins, are thought to be responsible for the health benefits that have traditionally been attributed to tea, especially green tea. The most active and abundant catechin in green tea is epigallocatechin-3-gallate (EGCG). The active catechins and their respective concentrations in green tea infusions are listed in the table below.
|
Catechin in Green Tea Infusion |
Catechin Concentration |
Catechin Concentration |
|
Epigallocatechin-3-gallate (EGCG) |
117–442 |
25–106 |
|
Epigallocatechin (EGC) |
203–471 |
49–113 |
|
Epicatechin-3-gallate (ECG) |
17–150 |
4–36 |
|
Epicatechin (EC) |
25–81 |
6–19 |
Black tea contains much lower concentrations of these catechins than green tea. The extended oxidation of black tea increases the concentrations of thearubigins and theaflavins, two types of complex polyphenols. Among their many biological activities, the predominant polyphenols in green tea―EGCG, EGC, ECG, and EC―and the theaflavins and thearubigins in black teas have antioxidant activity. These chemicals, especially EGCG and ECG, have substantial free radical scavenging activity and may protect cells from DNA damage caused by reactive oxygen species. Tea polyphenols have also been shown to inhibit tumor cell proliferation and induce apoptosis in laboratory and animal studies. In other laboratory and animal studies, tea catechins have been shown to inhibit angiogenesis and tumor cell invasiveness. In addition, tea polyphenols may protect against damage caused by ultraviolet (UV) B radiation, and they may modulate immune system function. Furthermore, green teas have been shown to activate detoxification enzymes, such as glutathione S-transferase and quinone reductase, that may help protect against tumor development. Although many of the potential beneficial effects of tea have been attributed to the strong antioxidant activity of tea polyphenols, the precise mechanism by which tea might help prevent cancer has not been established.
More than 50 epidemiologic studies of the association between tea consumption and cancer risk have been published since 2006. The results of these studies have often been inconsistent, but some have linked tea consumption to reduced risks of cancers of the colon, breast, ovary, prostate, and lung. The inconsistent results may be due to variables such as differences in tea preparation and consumption, the types of tea studied (green, black, or both), the methods of tea production, the bioavailability of tea compounds, genetic variation in how people respond to tea consumption, the concomitant use of tobacco and alcohol, and other lifestyle factors that may influence a person’s risk of developing cancer, such as physical activity or weight status. Several clinical trials have investigated the role of tea and tea polyphenols in cancer prevention. However, few trials have examined the effects of tea or tea polyphenols on cancer incidence or mortality. Two randomized trials evaluated the effects of tea extracts on premalignant oral lesions. One trial suggested a possible protective effect of tea on the development of oral cancer. In contrast, in the second trial no differences in lesion responses or histology were found between the groups. Two other randomized trials examined the effects of tea on urine levels of 8-hydroxydeoxyguanosine (8-OHdG), a biomarker of oxidative DNA damage that may be a predictor of increased cancer risk. Compared with those in the placebo group, individuals who took the green tea supplement at either dose for 3 months had substantially lower urinary 8-OHdG levels. Although these trials indicate that green tea polyphenols from tea or supplement can reduce urinary 8-OHdG levels, it is unclear if reduced 8-OHdG levels are associated with reduced cancer risk.
_
Coffee Consumption and Risk of Liver Cancer: A Meta-Analysis, 2007:
Four cohort and 5 case–control studies, involving 2260 cases and 239,146 noncases, met the inclusion criteria. All studies observed an inverse relation between coffee consumption and risk of liver cancer, and in 6 studies the association was statistically significant. Overall, an increase in consumption of 2 cups of coffee per day was associated with a 43% reduced risk of liver cancer (RR, 0.57; 95% CI, 0.49–0.67). There was no statistically significant heterogeneity among studies (P .17). In stratified analysis, the summary RRs of liver cancer for an increase in consumption of 2 cups of coffee per day were 0.69 (95% CI, 0.55–0.87) for persons without a history of liver disease and 0.56 (95% CI, 0.35–0.91) for those with a history of liver disease. Findings from this meta-analysis suggest that an increased consumption of coffee may reduce the risk of liver cancer.
_
Green (unfermented) tea, Camellia sinensis, has a reputation for being healthy and reducing cancer risk. Limited preclinical and clinical data seem to support this belief. In particular, a positive effect on the incidence of hepatocellular cancer was reported. However, a recent Cochrane Review found no evidence for a cancer-prevention effect of green tea. Interestingly, green tea consumption may contribute to weight loss or weight stabilisation. Therefore, an indirect effect of green tea on cancer incidence, mediated by its supposed weight-lowering property, cannot be excluded.
Like green tea consumption, regularly drinking coffee has been associated with a reduced risk for hepatocellular cancer. These findings are supported by two meta-analyses of several recent case-control and cohort studies. Intriguingly, consumption of more than 3 cups of coffee per day was also associated with an improved virologic response to peginterferon and ribavirin in patients with hepatitis C. This finding might provide a causal link between coffee and reduced hepatocellular cancer risk. However, there are no randomised controlled data on this issue. A meta-analysis of 13 cohort studies found no correlation between coffee consumption (more than 6 cups per day) and colorectal cancer risk. The same analysis found a small positive correlation (relative risk = 1.28) between tea consumption (more than 4 cups per day) and colorectal cancer incidence. However, relatively few participants of the study consumed that much tea, and the type of tea consumed (black versus green) was not assessed. If there is a real effect of coffee or tea on cancer risk, it must be small and would be difficult to detect in epidemiological studies.
_______
_______
Over Consumption of Calories and Cancer:
Eating too much food is one of the main risk factors for cancer. This can be shown two ways: (1) by the additional risks of malignancies caused by obesity, and (2) by the protective effect of eating less food.
Obesity has reached epidemic proportions in the United States. Sixty-four percent of the adult population is overweight or obese. About 1 in 50 are now severely obese (BMI > 40 kg/m2). Mokdad et al found that poor diet and physical inactivity was the second leading cause of death (400,000 per year in the USA), and would likely overtake tobacco as the leading cause of death.
_
It was estimated in a recent study, from a prospective cancer prevention cohort, that overweight and obesity accounted for 14 percent of all cancer deaths in men and 20 percent of those in women. Significant positive associations were found between obesity and higher death rates for the following cancers: esophagus, colon and rectum, liver, gallbladder, pancreas, kidney, stomach (in men), prostate, breast, uterus, cervix, and ovary. The authors estimated that over 90,000 cancer deaths per year could be avoided if the adult population all maintained a normal weight (BMI < 25.0). Clearly, obesity is a major risk factor for cancer.
_
Nearly all of the evidence linking obesity to cancer risk comes from large cohort studies, a type of observational study. However, data from observational studies cannot definitively establish that obesity causes cancer. That is because people with obesity or overweight may differ from people without these conditions in ways other than their body fat, and it is possible that these other differences—rather than their body fat—explain their increased cancer risk.
_
A nationwide cross-sectional study using BMI and cancer incidence data from the US Cancer Statistics database estimated that each year in 2011 to 2015 among people ages 30 and older, about 37,670 new cancer cases in men (4.7%) and 74,690 new cancer cases in women (9.6%) were due to excess body weight (overweight, obesity, or severe obesity). The percentage of cases attributed to excess body weight varied widely across cancer types and was as high as 51% for liver or gallbladder cancer and 49.2% for endometrial cancer in women and 48.8% for liver or gallbladder cancer and 30.6% for esophageal adenocarcinoma in men.
Globally, a 2019 study found that in 2012, excess body weight accounted for approximately 3.9% of all cancers (544,300 cases), with the burden of these cancer cases higher for women (368,500 cases) than for men (175,800 cases). The proportion of cancers due to excess body weight varied from less than 1% in low-income countries to 7% or 8% in some high-income Western countries and in Middle Eastern and Northern African countries.
_
An International Agency for Research on Cancer (IARC) Working Group concluded that there is consistent evidence that higher amounts of body fat are associated with an increased risk of a number of cancers. The table below shows the risks reported in representative studies.
|
Cancer type (reference) |
Compared with people without obesity or overweight, this cancer is |
|
Endometrial |
7 times as likely in people with severe obesity* |
|
Esophageal adenocarcinoma |
4.8 times as likely in people with severe obesity |
|
Gastric cardia |
2 times as likely in people with obesity |
|
Liver |
2 times as likely in people with obesity or overweight |
|
Kidney |
2 times as likely in people with obesity or overweight |
|
Multiple myeloma |
1.1–1.2 times as likely in people with obesity or overweight |
|
Meningioma |
1.5 times as likely in people with obesity |
|
Pancreatic |
1.5 times as likely in people with obesity or overweight |
|
Colorectal |
1.3 times as likely in people with obesity |
|
Gallbladder |
1.6 times as likely in people with obesity |
|
Breast |
0.8 times as likely in people with obesity or overweight |
|
Ovarian*** |
1.1 times as likely for every 5-unit increase in BMI |
|
Thyroid |
1.3 times as likely in people with obesity |
|
BMI = body mass index. |
|
People who have a higher BMI at the time their cancer is diagnosed or who have survived cancer have higher risks of developing a second, unrelated cancer (a second primary cancer).
_
Several possible mechanisms have been suggested to explain how obesity might increase the risks of some cancers.
- Fat tissue (also called adipose tissue) produces excess amounts of estrogen, high levels of which have been associated with increased risks of breast, endometrial, ovarian, and some other cancers.
- People with obesity often have increased blood levels of insulin and insulin-like growth factor-1 (IGF-1). High levels of insulin, a condition known as hyperinsulinemia, is due to insulin resistance and precedes the development of type 2 diabetes, another known cancer risk factor. High levels of insulin and IGF-1 may promote the development of colon, kidney, prostate, and endometrial cancers. Calle and Kaaks proposed that insulin and insulin-like growth factor 1 (IGF-1) are pathways that are linked to obesity and work to prohibit apoptosis and promote cell proliferation. Hyperinsulinemia triggers the insulin-IGF pathway, a complex pathway that includes insulin, IGF-1, and IGF-2 (three ligands), along with six receptors (insulin receptor α, IR β, IGF-1 receptor, IGF-2R, hybrid IGF-1R/IR α, and hybrid IGF-1R/IR β) and the seven IGF-binding proteins. It is hypothesized that the increased level of insulin reduces the amount of IGFBPs, which leads to an increase in the level of IGF-1 and a change in cell environment which promotes tumor growth. Growth hormone, which is regulated by insulin, stimulates production of IGF-1 and IGFBP-3. The hypothesized pathways have been demonstrated within in vitro and in vivo (animal) studies. In meta-analyses, an increased level of IGF-I has been associated with increased risk of pre and postmenopausal breast cancers and prostate cancer.
- People with obesity often have chronic inflammatory conditions such as gallstones or non-alcoholic fatty liver disease. These conditions can cause oxidative stress, which leads to DNA damage and increases the risk of biliary tract and other cancers. Substantial evidence has been shown between obesity, chronic inflammation, and insulin resistance. Based on previous research on obesity-related inflammation and insulin resistance, researchers have hypothesized and extrapolated the relationship to include tumorigenesis. However, there is no strong evidence.
- Fat cells produce hormones called adipokines that can stimulate or inhibit cell growth. For example, the level of an adipokine called leptin in the blood increases with increasing body fat, and high levels of leptin can promote aberrant cell proliferation. Another adipokine, adiponectin, is less abundant in people with obesity than in people with a healthy weight and may have antiproliferative effects that protect against tumor growth.
- Fat cells may also have direct and indirect effects on other cell growth and metabolic regulators, including mammalian target of rapamycin (mTOR) and AMP-activated protein kinase.
- Recently, Roberts and colleagues provided a new perspective on the biological mechanisms by which obesity may influence cancer by highlighting obesity-related hypoxia, shared genetic susceptibility, migrating adipose stromal cells, and other biological candidates (obesity related inflammation, oxidative stress, and nuclear factor kB system). They point to the relationship between adipose tissue hypoxia (ATH) and the development of insulin resistance that leads to reduction of adiponectin and increased leptin gene expressions. The in vivo study showed there may be lower oxygen levels in obese mice than lean mice. The in vitro studies found that melanoma grew more readily in a hypoxic microenvironment. With the improvement in technology and accelerated pace in genome-wide association studies, obesity genetics and its relationship with cancer genes are widely studied within breast and colorectal cancers. When the obesity gene map is superimposed with cancer gene maps there seem to be connections with chromosomes 11p and 16q for breast cancer and 18q for colorectal cancer.
Other possible mechanisms by which obesity could affect cancer risk include impaired tumor immunity and changes in the mechanical properties of the scaffolding tissue that surrounds developing tumors.
In addition to biological effects, obesity can lead to difficulties in screening and management. For example, women with overweight or obesity have an increased risk of cervical cancer compared with women of healthy weight, likely due to less effective cervical cancer screening in these individuals.
_____
Calories restriction and cancer:
On the other side, careful menu planning brings about an approach entitled CRON-Calorie Restriction with Optimal Nutrition. The basic idea is to eat a reduced amount of food (about 70–80 percent of the amount required to maintain “normal” body weight) while still consuming all of the necessary amounts of vitamins, minerals, and other necessary nutrients. The only restriction is the total amount of energy (calories) that is consumed. While being difficult to practice, this approach has a lot of scientific merit for being able to extend average life spans of many species of animals including rats, mice, fish, and possibly primates (currently being tested). Along with this life span extension is a reduction in chronic diseases that are common to mankind, reviewed in Hursting et al. A recent meta-analysis of 14 experimental studies found that energy restriction resulted in a 55% reduction in spontaneous tumors in laboratory mice. Calorie restriction inhibited induced mammary tumors in mice and suppressed implanted tumor growth and prolonged survival in energy restricted mice. Among Swedish women who had been hospitalized for anorexia nervosa (definitely lower caloric intake, but not adequate nutrition) prior to age 40, there was a 23% lower incidence of breast cancer for nulliparous women and a 76% lower incidence for parous women. So, too many calories is definitely counter-productive, and slightly less than normal is very advantageous.
_
Calorie restriction reduces the exposure of cells to high glucose levels, insulin and other growth factors, favouring a metabolic state dominated by AMPK, while at the same time inhibiting mTOR activity. It is no surprise, therefore, that calorie reduction does lower the frequency of spontaneous and induced tumours in laboratory animals.
For obvious reasons, there is only circumstantial evidence that drastic calorie reduction has the same effect in humans. The example most often cited is a study of the population of Okinawa with a total energy intake 20% below the Japanese (and 40% below the US) average. They live longer, and death rates due to cancer are 30% lower than in the rest of Japan. Another example is provided by individuals who experienced the “Dutch Hunger Winter” during World War II (1944–1945) and who had a lower risk of developing colorectal cancer later in life. In this case, unvoluntary severe CR, although of short duration, led to epigenetic changes that might explain the outcome.
It is impossible to impose substantial (20–40%) energy reduction regimes on human populations for a prolonged period of time. Healthy people subjected to this kind of experiment do not get used to feeling hungry, become apathic, cold-sensitive and sometimes depressive, as Benedict et al. described in a careful, book-length study published in 1919. It might even be dangerous, as discussed by Dirks et al. and by Mattson who speculated, based on single observations and some animal experiments, that a causal link between a negative energy balance and the motor neuron disease Amyotrophic Lateral Sclerosis (ALS) might exist.
Diets calorie-reduced only by about 10% should be feasible and have been shown to improve certain risk factors. However, no large long-term study on the effect of such a diet on cancer incidence has been published.
_
Does losing weight of obese patient lower the risk of cancer?
Most of the data about whether losing weight reduces cancer risk comes from cohort and case–control studies. Observational studies of obesity and cancer risk should be interpreted with caution because they cannot definitively establish that obesity causes cancer and people who lose weight may differ in other ways from people who do not.
Some of these studies have found decreased risks of breast, endometrial, colon, and prostate cancers among people with obesity who had lost weight. For example, in one large prospective study of postmenopausal women, intentional loss of more than 5% of body weight was associated with lower risk of obesity-related cancers, especially endometrial cancer. However, unintentional weight loss was not associated with cancer risk in this study.
A follow-up study of weight and breast cancer in the Women’s Health Initiative found that, for women who were already overweight or obese at the beginning of the study, weight change (either gain or loss) was not associated with breast cancer risk during follow-up. However, in a study that pooled data from 10 cohorts, sustained weight loss was associated with lower breast cancer risk among women 50 years and older.
To better understand the relationship between weight loss among people with obesity and cancer risk, some researchers are examining cancer risk in people with obesity who have undergone bariatric surgery. Studies have found that bariatric surgery among people with obesity, particularly women, is associated with reduced risks of cancer overall; of hormone-related cancers, such as breast, endometrial, and prostate cancers; and of obesity-related cancers, such as postmenopausal breast cancer, endometrial cancer, and colon cancer.
_____
_____
Section-8
Infectious causes of Cancer:
Worldwide, approximately 20 % of cancer cases are related to infectious diseases. This proportion varies in different regions of the world from a high of 25% in Africa to less than 10% in the developed world. Viruses are the usual infectious agents that cause cancer but bacteria and parasites also contribute. Infectious organisms that increase the risk of cancer are frequently a source of DNA damage or genomic instability.
_
Viruses:
Viral infection is a major risk factor for cervical and liver cancer. A virus that can cause cancer is called an oncovirus. Approximately 20% of all cancers are associated with infectious agents, and 12% of all cancers are caused by oncoviruses. 80% of viral cancers occur in the developing world. Oncovirus infections are common, but these infections rarely result in cancer. One or more additional insults, such as chronic inflammation, environmental mutagens, or immunosuppression, are required for cancer development. Additionally, viruses are only an absolute requirement for oncogenesis in Kaposi sarcoma and cervical cancer. Oncoviruses are classified as direct or indirect carcinogens, although some overlap exists between the distinctions. Direct carcinogenic viruses possess viral oncogenes that directly contribute to neoplastic cellular transformation, whereas indirect carcinogens cause chronic inflammation, which can lead to oncogenic transformation. Oncogenic DNA viruses include EBV, hepatitis B virus (HBV), human papillomavirus (HPV), human herpesvirus-8 (HHV-8), and Merkel cell polyomavirus (MCPyV). Oncogenic RNA viruses include, hepatitis C virus (HCV) and human T-cell lymphotropic virus-1 (HTLV-1).
_
Viral cancers do not arise acutely after infection, but instead develop 15–40 years later. One exception is a rare EBV-associated lymphoproliferative disease, which can occur shortly after infection. In cancers, viral replication is either diminished or absent, as active replication would lyse the host cell and prevent tumorigenesis. The virus exists intracellularly as naked nucleic acid in the form of a plasmid, episome, or cellular-integrated genome. DNA virus genomes can integrate directly into the host genome, while RNA virus genomes must undergo reverse transcription to DNA before integration can occur.
_
In general, all oncoviruses promote tumorigenesis via common pathways. Tumor suppressor pathways, such as p53 and retinoblastoma (Rb), are often inhibited. Other frequent targets include, tumor necrosis-associated factors (TRAFs), telomerase reverse transcriptase (TERT), cytoplasmic PI3K-AKT-mTOR, nuclear factor-κB (NF-κB), β-catenin, interferon signaling pathways, major histocompatibility class-1 (MHC-1), and Janus kinase/signal transducer and activator of transcription (JAK/STAT). The host DNA damage response pathway (DDR) can also be affected, particularly by DNA viruses. The DDR detects and repairs damaged cellular DNA via responses, initiated by the phosphoinositide-3-like kinase (PIKK) family of serine/threonine kinases, including ataxia-telangiectasia mutated (ATM), ataxia-telangiectasia and RAD3-related (ATR), and DNA-dependent protein kinase (DNA-PK). Cell cycle progression may be delayed by the DDR until DNA repair is completed or foreign viral DNA is no longer detected. To promote oncogenesis, viral proteins activate aspects of the DDR that are beneficial to viral replication or cellular transformation, such as repair factor recruitment, and inactivate DDR activities that are detrimental to viral DNA survival, such as apoptotic pathways. The oncogenic potential of some viruses has been clearly established, and viruses are thus becoming targets for cancer treatment and prevention. Successful vaccines are already available for HBV and HPV infection prevention.
_
In Western developed countries, human papillomavirus (HPV), hepatitis B virus (HBV) and hepatitis C virus (HCV) are the most common oncoviruses. In the United States, HPV causes most cervical cancers, as well as some cancers of the vagina, vulva, penis, anus, rectum, throat, tongue and tonsils. Among high-risk HPV viruses, the HPV E6 and E7 oncoproteins inactivate tumor suppressor genes when infecting cells. In addition, the oncoproteins independently induce genomic instability in normal human cells, leading to an increased risk of cancer development. Individuals with chronic hepatitis B virus infection are more than 200 times more likely to develop liver cancer than uninfected individuals. Liver cirrhosis, whether from chronic viral hepatitis infection or excessive alcohol use, is independently associated with the development of liver cancer, but the combination of cirrhosis and viral hepatitis presents the highest risk of liver cancer development.
_
DNA oncoviruses typically impair two families of tumor suppressor proteins: tumor proteins p53 and the retinoblastoma proteins (Rb). It is evolutionarily advantageous for viruses to inactivate p53 because p53 can trigger cell cycle arrest or apoptosis in infected cells when the virus attempts to replicate its DNA. Similarly, Rb proteins regulate many essential cell functions, including but not limited to a crucial cell cycle checkpoint, making them a target for viruses attempting to interrupt regular cell function. Rb and p53 regulate the transition between G1 and S phase, arresting the cell cycle before DNA replication until the appropriate checkpoint inputs, such as DNA damage repair, are completed. p53 regulates the p21 gene, which produces a protein which binds to the Cyclin D-Cdk4/6 complex. This prevents Rb phosphorylation and prevents the cell from entering S phase. In mammals, when Rb is active (unphosphorylated), it inhibits the E2F family of transcription factors, which regulate the Cyclin E-Cdk2 complex, which inhibits Rb, forming a positive feedback loop, keeping the cell in G1 until the input crosses a threshold. To drive the cell into S phase prematurely, the viruses must inactivate p53, which plays a central role in the G1/S checkpoint, as well as Rb, which, though downstream of it, is typically kept active by a positive feedback loop. DNA oncoviruses typically cause cancer by inactivating p53 and Rb, thereby allowing unregulated cell division and creating tumors. RNA virus genomes must undergo reverse transcription to DNA before integration into host genome can occur.
_
Bacteria and parasites:
Certain bacterial infections also increase the risk of cancer, as seen in Helicobacter pylori-induced gastric carcinoma. The mechanism by which H. pylori causes cancer may involve chronic inflammation or the direct action of some of the bacteria’s virulence factors. Parasitic infections strongly associated with cancer include Schistosoma haematobium (squamous cell carcinoma of the bladder) and the liver flukes, Opisthorchis viverrini and Clonorchis sinensis (cholangiocarcinoma). Inflammation triggered by the worm’s eggs appears to be the cancer-causing mechanism. Certain parasitic infections can also increase the presence of carcinogenic compounds in the body, leading to the development of cancers. Tuberculosis infection, caused by the mycobacterium M. tuberculosis, has also been linked with the development of lung cancer.
_____
_____
Summary of microbial factors and their mechanisms inducing carcinogenesis:
|
Microbial signature |
Cancer type |
Mediators |
Mechanism |
|
Helicobacter pylori |
Gastric cancer, Mucosal associated lymphoid tissue lymphoma, Colorectal cancer, Pancreatic cancer |
Adhesin molecules- BabA, SabA, OipA, AlpA/B; Virulence effectors- CagA, VacA, OMPs |
Binds to host cell surface molecules, acts of SHP2 tyrosine phosphatase, and increased expression of cell proliferation genes and pathways (RAS-MAPK); Stimulates IL-8, IL-12 mediated inflammatory reactions; Vac A upon internalization promotes expression of cell cycle factors, loss of mitochondrial membrane integrity and upregulation of autophagy |
|
Fusobacterium nucleatum |
Colorectal cancer |
Virulence factors- FadA, Fap2 |
Interacts with cadherin group of adhesion molecules on the host cell surface, internalization and activation of TLR4 signaling, enhanced β catenin translocation and oncogene expression; Promotion of TLR4/MYD88/NF-kB, RAS-MAPK signaling cascades, expression of ncRNAs (miR-21) and recruitment of myeloid-derived cells; ROS production and hypermethylation of repair machinery; Fap2 interacts with inhibitory domains on the surface of immune cells and suppresses immune functions |
|
Esophageal cancer |
|||
|
Bacteroides fragilis |
Colorectal cancer |
Toxin-BFT |
Binds to and activates the STAT3 transcription factor which works either by promoting the inflammatory cascade or by ROS production and DNA damage; PGE2 activates PI3K-AKT and inhibits apoptosis, RAS-MAPK pathway and promotes proliferation, suppresses immune function by inhibiting TH1 and upregulating TH2 activities; Increased production of proinflammatory cytokines such as IL-6, IL-17, TNFα |
|
Escherichia coli |
Colorectal cancer, Esophageal adenocarcinoma |
Secondary metabolite- colibactin |
Binds to the DNA minor groove, alkylates, and generates interstrand crosslinks, DSBRs, ROS production, and cell cycle arrest |
|
Salmonella typhi |
Gall bladder cancer, colorectal adenocarcinoma |
Extracellular polymeric substances, Cytolethal distending toxin B, cytotoxic necrotizing factor 1 |
Toxins bind to and impose DNA damaging effects and also promote inflammation |
|
Chlamydophila pneumoniae |
Lung cancer |
Oncogenic proteins |
Perturbations in the mitochondrial functions, cellular activities, apoptotic events; Cytokine release and chronic inflammation, evasion of immune responses through apoptotic polymorphic neutrophils |
|
Porphyromonas gingivalis |
Oral squamous cell carcinoma Pancreatic cancer Esophageal cancer |
Nucleoside diphosphate kinase, Gingipains |
Invades and resides in the gingival epithelial cells; Overexpression of B7-H1 and B7-DC receptors and enhanced production of TNF-α, IL-1, IL-6, and IL-8; Increased expression of pro-MMP-9, MMP-10, MMP-1 and aids in EMT; Phosphorylation and activation of CDKs thus inducing cellular proliferation, inhibition of TP53 and apoptosis through activation of JAK1/STAT3, PI3K/Akt signaling pathway |
|
Human papilloma virus |
Cervical cancer |
Oncoproteins-E6, E7 |
Maintained in the episomal state E7 inhibits terminal differentiation of epithelial cells and pRb which sets E2f free thus promoting the expression of cell proliferation genes; E6 sequesters p53, degrades it, inhibits apoptosis via NF-kβ signaling, interacts with myc, NFX1-91 and sustains telomerase in an activated state; Downregulates host innate immune responses by repressing TLR9 signaling |
|
Oral cancer |
|||
|
Head and neck cancer |
|||
|
Hepatitis B virus |
Hepatocellular carcinoma |
Oncoprotein- Hepatitis BX (HBx) |
Interacts with the promoters of cell proliferation genes such as NF-kβ, IL-8, TNF, EGFR, Jak1-STAT; HBXIP binds with centrosome regulator, perturbs centrosome duplication, generation of multipolar mitotic spindle formation; Associates and inhibits p53, suppresses apoptosis, promotes angiogenesis, expression of DNA methyltransferases thus inhibiting the activities of tumor suppressor gene |
|
Hepatitis C virus |
Hepatocellular carcinoma |
Oncoprotein NS5A |
Activates PI3K/Akt, Wnt-β cell proliferation pathways inhibits the cell cycle negative regulator CDKN1A; Proliferation is also enhanced by the interaction with RXR, PPAR α, promotes fatty acid transport and catabolism, maintains telomere lengths |
|
Epstein Barr virus/Human Herpesvirus Type 4 |
Hodgkin’s Lymphoma |
Oncoprotein- LMP1, LMP2A |
LMP1 activates STAT, NF-kB transcription factors MAP kinases, PI3K pathways |
|
Burkitt’s Lymphoma |
LMP2A promotes EMT by activation of PLCγ and PI3K pathways |
||
|
Human T-Lymphotropic Virus Type 1 |
Adult T-cell leukemia |
Oncoprotein- Tax |
Reprogramming of cell cycle events and DNA repair inhibition; Induction of NF-kβ pathway, upregulation of cytokine expression, IL-2, IL-13, IL-15 leading to chronic inflammatory reactions |
|
Kaposi sarcoma-associated herpesvirus |
Kaposi sarcoma |
Oncoprotein- Latency-associated nuclear antigen (LANA), K1, K3, K5, v-Cyclin, v-FLIP, v-Bcl2, v-IRF |
Cell cycle regulation by LANA 1 and v-Cyclin K1, v-Bcl-2, and v-FLIP inhibits apoptosis; K3, K5, and v-IRF exert immune modulatory effects |
|
Schistosoma haematobium |
Urinary bladder cancer |
Nitrosamines |
Initiates chronic inflammatory reactions, generation of reactive oxygen species, DNA damage, inhibition of p53, Rb |
|
Schistosoma japonicum |
Hepatocellular carcinoma |
Stimulates chronic inflammation, mutations in p53 gene, genomic instability |
|
|
Colon cancer |
|||
|
Metastatic Lung cancer |
|||
|
Schistosoma mansoni |
Hepatocellular carcinoma |
Inhibits cell-mediated immune responses |
|
|
Schistosoma mekongi |
Leiomyosarcoma of small bowel |
||
|
Schistosoma intercalatum |
Rectosigmoid carcinoma |
||
|
Urinary bladder cancer |
|||
|
Opisthorchis viverrini |
Cholangiocarcinoma |
Dimethylnitrosamine, Granulin, Thioredoxin peroxidase |
Biliary cell proliferation, inhibition of apoptosis; Activation of PI3K/Akt and Wnt/β-catenin pathways, cytokine production; Inhibition of Rb1 and p16, and promotion of cyclin D1 CDK4 |
|
Clonorchis sinensis |
Cholangiocarcinoma |
8-oxodG |
Promotes squamous metaplasia, mucous gland hyperplasia, inflammatory responses, and DNA damage |
_____
_____
Viruses and Human Cancers: A Long Road of Discovery of Molecular Paradigms, 2014 study:
About a fifth of all human cancers worldwide are caused by infectious agents. In 12% of cancers, seven different viruses have been causally linked to human oncogenesis: Epstein-Barr virus, hepatitis B virus, human papillomavirus, human T-cell lymphotropic virus, hepatitis C virus, Kaposi’s sarcoma herpesvirus, and Merkel cell polyomavirus. Here, authors review the many molecular mechanisms of oncogenesis that have been discovered over the decades of study of these viruses. They discuss how viruses can act at different stages in the complex multistep process of carcinogenesis. Early events include their involvement in mutagenic events associated with tumor initiation such as viral integration and insertional mutagenesis as well as viral promotion of DNA damage. Also involved in tumor progression is the dysregulation of cellular processes by viral proteins, and authors describe how this has been investigated by studies in cell culture and in experimental animals and by molecular cellular approaches. Also important are the molecular mechanisms whereby viruses interact with the immune system and the immune evasion strategies that have evolved.
_
The issue of establishing causality:
First, it is often the case that there is a long latency period between primary viral infection and occurrence of the cancer. For example, the latency period between HTLV-1 infection and onset of acute T-cell leukemia is on the order of decades, and only a minor fraction of individuals who are infected will go on develop ATL. Similarly, virus infection can often be subclinical, and so it is difficult to establish the time of infection. For many cancer viruses, infection is widespread but the associated cancer is rare. For example, seroepidemiological studies show that 63 to 75% of the population has been exposed to MCV, but the incidence of Merkel cell carcinoma (MCC) is 0.17/100,000 to 0.34/100,000 in the United States. Some cancers often require cofactors as well as the virus to develop. For example, in the case of HPV, cofactors in the development of cervical cancer include smoking, hormonal contraceptives, nutrition, and coinfections with other organisms, such as herpesvirus, Chlamydia, and HIV. In some cancers, the virus may integrate irreversibly into the host genome during pathogenesis (e.g., with MCV), so it is impossible to culture infectious progeny. The outcome of virus infection may vary depending on host factors such as immune status; e.g., the impaired immune system in HIV/AIDS is a major predisposing factor for Kaposi’s sarcoma. Viruses may employ different mechanisms in the multistage process of carcinogenesis: HPV promotes chromosomal instability and so may directly contribute to cellular genetic changes, whereas the role of HBV and HCV in HCC development is more indirect, involving chronic inflammatory responses and taking many decades while mutations accumulate, often aided by exposure to aflatoxin and alcohol. Finally, for many viruses there is a lack of an animal model, and indeed some, e.g., MCV, lack even a cell culture system. It should also be noted that viruses can cause cancers with histopathological features similar to those of cancers caused by other factors.
_
In order to address these problems, a number of approaches have been suggested, such as criteria defining environmental causes, consistency, specificity, temporality, plausibility, etc., and epidemiological approaches. Guidelines have been proposed for relating a given virus to a human cancer. Briefly, these are as follows: (i) the geographical distribution of viral infection should match that of cancer after adjustment for other cofactors. (ii) viral markers (e.g., antiviral antibody titer or presence of virus-specific cytotoxic T cells) should be higher in cases of cancer than in controls, (iii) viral markers being present should precede the tumor and have an incidence that matches the incidence of the tumor, (iv) prevention of viral infection (e.g., by vaccination) should decrease incidence of the tumor, (v) the virus should exhibit transforming properties with human cells in culture, and (vi) the virus should induce tumors in animals and this should be preventable by viral neutralization. Recently, humanized mouse models have been developed to study viral infections, e.g., for HBV and HCV and EBV. In practice, these issues can be complex and depend on the virus. For example, there is an increased risk of certain types of noninfectious cancers in HIV-1-infected individuals, presumably due to the defective cell-mediated immunity, but HIV-1 is not considered an oncovirus. Clearly, much research into the epidemiology, virology, and molecular biology of a virus is needed before it can be accepted as an oncovirus.
_
Viral mechanisms of transformation and tumorigenicity:
Investigation of the molecular mechanisms used by the viral transforming proteins, including those of the seven human oncoviruses, is still an active area of research today.
Human cancer viruses: transformation and tumorigenicity:
|
Virus |
Abbreviation(s) |
Type |
Family |
Cancer(s) |
Tropism(s) |
Integration |
Infectious virus produced |
Oncoproteins |
|
Human papillomaviruses 16 and 18 |
HPV16, HPV18 |
DNA |
Papillomavirus |
Cervical carcinoma, anal carcinoma, penile carcinoma |
Keratinocytes |
Episomal and integrated |
Yes |
E6, E7 |
|
Epstein-Barr virus |
EBV, HHV-4 |
DNA |
Herpesvirus |
Burkitt’s lymphoma, nasopharyngeal carcinoma |
B lymphocytes, epithelial cells |
Episomal |
Yes |
EBNA-1, EBNA-2 LMP-1, LMP-2 EBERs, BARTs |
|
Kaposi’s sarcoma-associated herpesvirus |
KSHV, HHV-8 |
DNA |
Herpesvirus |
Kaposi’s sarcoma, primary effusion lymphoma, multicentric Castleman’s disease |
B cells, macrophages, keratinocytes, endothelial cells, etc. |
Episomal |
Yes |
LANA, vIL-6, vMIP-I, vFLIP, vBCL-2, v-cyclin-D, vGPCR, vIRF-1 |
|
Hepatitis B virus |
HBV |
DNA |
Hepadnavirus |
Hepatocellular carcinoma |
Liver |
Yes |
Yes |
HBx, HBsAg |
|
Hepatitis C virus |
HCV |
RNA |
Flavivirus |
Hepatocellular carcinoma |
Liver |
No |
Yes |
Core protein, NS3 |
|
Human adult T-cell leukemia virus type 1 |
HTLV-1 |
RNA |
Retrovirus |
T-cell leukemia |
T lymphocytes |
Yes |
Yes |
Tax, p12, p30 p8, p13 |
|
Merkel cell polyomavirus |
MCV, MCPyV |
DNA |
Polyomavirus |
Merkel cell carcinoma |
Merkel cells |
Yes |
No |
T antigen, t antigen |
_
In general, cancer development is a complex multistep process, and this is also true for cancers arising from the effects of viruses, which exert effects on different stages of tumor formation depending on the virus. Viruses can exert their effects on the early stage of oncogenesis involving genetic events that are involved in tumor initiation or, alternatively, act at later stages by modulating signaling pathways involved in the regulation of cell proliferation, apoptosis, replicative immortality, and others, such as tumor promotion.
_
Several human oncoviruses, including HPV, can cause genetic changes to the host genome that can initiate and contribute to carcinogenesis. In the case of HPV, the viral genome is normally episomal in basal epithelial cells that have become infected by virus but are not producing virus. However, in HPV-transformed cells, i.e., those that are infected and have begun malignant progression, it often becomes integrated into the host DNA by random integration events which leave the E6 and E7 oncogenes (see below) intact and still expressed, underlining their importance in tumorigenesis. Integration is not a normal part of the life cycle of HPV, and when it occurs, HPV becomes unable to complete its life cycle. Indeed, there is evidence that both episomal and integrated HPV can be present at the same time during the early phase of tumorigenesis and that episomal HPV become less common at later stages, but there is continued and robust expression of E6 and E7. E6 and E7 are thought to have a role in carcinogenesis for the high-risk HPVs (HPV16, -18, and -31). Cells with integrated HPV proliferate more rapidly and form a pool of immortalized cells where it is possible for further mutations to occur and lead to carcinoma formation. Enhancing this, E6 and E7 cause genetic instability, and it has been reported that acquirement of elevated levels of chromosomal instability is associated with the integration of HPV16 in cervical keratinocyte cell lines.
_
The oncogenic properties of the high-risk HPVs (HPV16, -18, and -31) are thought to involve mainly the proteins E6 and E7. As shown in figure below, cellular p53 is a target of E6, and E6 binds and degrades p53 through an E3 ubiquitin protein ligase called cellular E6-associated protein (E6-AP), which results in the ubiquitination and degradation of p53. E6 also binds p300/CBP, which is a p53 coactivator, resulting in indirect interference with p53, binds and inactivates p21 and p27, hDlg, MUPP1, and hScrib, and may also affect Notch signaling and activate the hTERT promoter via an E6/Myc/Max complex, bind proapoptotic Bak, and promote phosphorylation of pRb.
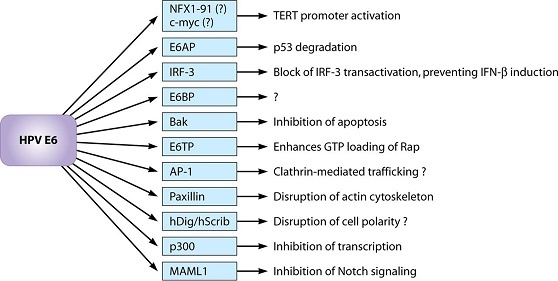
Figure above shows schematic representation of signaling by high-risk human papillomavirus HPV E6. Targets for HPV E6 (center column) and biological effects (right column) are shown. E6AP, E6AP ubiquitin-protein ligase (UBE3A); IRF-3, interferon-regulatory transcription factor 3; E6BP, E6 binding protein; E6TP, E6-targeted protein 1; AP-1, activator protein 1; hDig/hScrib, human homolog of Drosophila disc-large tumor suppressor/human homolog of Drosophila Scribble; p300, p300 transcriptional coactivating protein; MAML1, human homolog of Drosophila mastermind; TERT, telomerase reverse transcriptase.
The other important protein in HPV carcinogenesis is E7, a phosphoprotein with two zinc fingers that shows some structural and functional similarity to adenovirus E1A and SV40 large T antigen. E7 can self-assemble into spherical oligomers and is phosphorylated by casein kinase II, and this is inhibited by myeloid-related protein 8 (MRP-8) and MRP-14, which are growth inhibitory and complex with pRb, p107, and p130, resulting in phosphorylation and E2F release, which promotes cell cycle progression. E7 can also bind histone deacetylases (HDACs), the S4 subunit of proteasome, Mi2β, AP-1, MPP2, TATA-binding protein (TBP), and hTid-1, and, like E6, it induces chromosomal instability and cooperates with E6 in this induction. Thus, the high-risk HPVs are powerful human cancer viruses that express oncoproteins that act at multiple stages in tumorigenesis from events involved in tumor initiation to later stages of tumor promotion.
_
The HBV HBx protein is oncogenic in experimental hepatocellular carcinogenesis, since it is able to transform rodent hepatocytes in vitro. HBx sequences persist in clonally expanding rodent hepatocytes, and HBx transforms NIH 3T3 cells in cooperation with Ras. Expression of the HBV protein HBx in transgenic mice makes them susceptible to chemical carcinogens, and high levels of HBx expression can lead to HCC. HBx acts as an oncogene in experimental HCC and accelerates HCC development in the presence of Myc in transgenic mice without cirrhosis and also augments their susceptibility to develop HCC after exposure to diethylnitrosamine (a strong carcinogen). Further, lowering the levels of HBx mRNA and protein by RNA interference results in a reduction in the tumorigenicity of HCC cells that constitutively express HBx. For HCV, in transgenic mice expressing HCV core protein, hepatic steatosis develops early in life and can go on to give liver adenomas and HCC. The 5′ half of the cDNA for HCV nonstructural protein 3 (NS3) can transform cells in culture and confer tumorigenicity in nude mice for NIH 3T3 cells, rat fibroblasts, and the QSG7701 human liver cell line. HBsAg expression in transgenic mice has also been reported to lead to HCC. HCV core protein transforms NIH 3T3 cells via STAT3 activation. The function of HBx is not yet well understood, but it may play a part in promoting viral transcription. HBx antigen is present mainly in the cytoplasm but also in the nucleus and, as shown in figure below, activates the extracellular signal-regulated kinase (ERKs), stress-activated protein kinases (SAPKs), and p38 protein kinase pathways. It has also been reported to bind to p53.
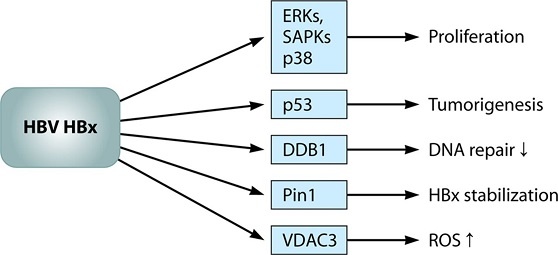
Figure above shows schematic representation of signaling by HBV HBx. Targets for HBV HBx (center column) and biological effects (right column) are shown. ERKs, extracellular signal-regulated kinases; SAPK, stress-activated protein kinase; DDB1, damage-specific DNA-binding protein 1; Pin1, peptidylprolyl cis/trans isomerase; VDAC3, voltage-dependent anion channel 3.
_
Viral mechanisms of immune evasion:
The human body has elaborated a complex variety of immune defense mechanisms that prevent viral infections and eliminate virally infected cells. In turn, viruses have adapted by evolving different strategies for evading immune responses. In general, it can be said that these mechanisms are also involved in the immune evasion of virally induced cancers and that this is not because the virus evolved to cause cancer but rather is a secondary result of the mechanisms that the virus deployed to evade the elimination of virally infected cells. Indeed, this is similar to the uncontrolled growth of virally induced tumors, which is a secondary result of the mechanisms that the virus evolved to enhance cell proliferation in order to enhance viral replication or perpetuate persistence of the viral genome. The interplay between virus and immune system is apparent from the observation that virally induced cancers, e.g., Kaposi’s sarcoma, Merkel cell carcinoma, and EBV-associated lymphomas, are more frequent in individuals with an impaired immune status. The mechanisms involved in interactions between human cancer viruses and the immune system are depicted in table below:
Human cancer viruses: mechanisms of immune evasion:
|
Virus |
Abbreviation(s) |
MHC regulation |
Interferon pathway change(s) |
Molecular mimicry |
Escape mutants |
|
Human papillomaviruses 16 and 18 |
HPV16, HPV18 |
LMP2 and LMP7 ↓, TAP1 and TAP2 ↓, MHC-I ↓ |
TLR9 ↓ |
||
|
Epstein-Barr virus |
EBV, HHV-4 |
TAP ↓ by BNLF2a, MHC-I ↓ by BNLF5, TAP1 ↓ by vIL-10, MHC-I ↓ by BILF1, MHC-II ↓ by gp42/gH/gL, BGLF5, and vIL-10 |
Tyk2↓ by LMP-1, TLR9 ↓ by LMP-1 |
||
|
Kaposi’s sarcoma-associated herpesvirus |
KSHV, HHV-8 |
MHC-I ↓ by MIR1 and MIR2 |
IFN signaling ↓ by RIF |
v-CCL-1, v-CCL-2, v-CCL-3, v-FLICE |
|
|
Hepatitis B virus |
HBV |
MHC-I ↓ |
IFN-α and IFN-β ↓ |
Env mimics IgA |
Yes |
|
Hepatitis C virus |
HCV |
RIG-1 ↓ by NS3/4A, TRIF ↓ by NS3/4A, IL-8 ↑ by NS5A |
Yes |
||
|
Human adult T-cell leukemia virus |
HTLV-1 |
TLR4 signaling ↓ by p30 |
|||
|
Merkel cell polyomavirus |
MCV, MCPyV |
TLR9 ↓ by T-Ag |
Other Strategies:
A variety of other strategies have been employed by human oncoviruses to avoid immune responses. One simple one is to just express proteins at a very low level: HTLV-1 downregulates the levels of viral gene expression but still persists through the proliferation of infected cells harboring the latent viral episome. HPV has evolved multiple strategies of immune evasion so as to persist in squamous epithelia for enough time to complete the viral replicative life cycle. This persistent HPV infection is the principal risk factor for developing HPV-associated precancerous lesions and cancers. Clearance of HPV-induced lesions involves cellular immune responses, and the interplay of these responses with viral immune evasion mechanisms determines whether HPV infection is cleared or persists. The virus is able to maintain a low profile, since it exclusively infects epithelial cells and its replicative cycle occurs outside the basement membrane away from immune effector cells. Also, viral proteins are not secreted but are expressed mainly at low levels in the nuclei of infected cells early in infection by the viral gene repressor protein E2 and usage of rare codons, which limits viral protein translation. During the later stages of infection, viral protein expression occurs only in the keratinocytes of the upper layers of the epithelium, where access by the immune system is limited, and indeed HPV infection does not involve viremia, cell death, or cell lysis upon viral shedding. In this way, HPV avoids activation of antigen-presenting cells, production of cytokines, and initiation of the immune response.
______
______
Chronic bacterial and parasitic infections and cancer: a review, 2010:
A relatively underestimated facet of infectious diseases is the association of chronic bacterial and parasitic infections with cancer development. Therefore, authors sought to evaluate the evidence regarding the association of such infections with the development of malignancy, excluding the overwhelming evidence of the association of Helicobacter pylori and cancer. Authors searched Pubmed, Cochrane, and Scopus without time limits for relevant articles. There is evidence that some bacterial and parasitic infections are associated with cancer development. The level of evidence of this association varies from high to low; in any case, a long time interval is mandatory for the development of cancer. A high level of evidence exists for the association of Salmonella Typhi with gallbladder and hepatobiliary carcinoma; Opisthorchis viverrini and Clonorchis sinensis with cholangiocarcinoma; Schistosoma hematobium with bladder cancer; chronic osteomyelitis with squamous cell carcinoma of the skin; and hidradenitis suppurativa with squamous cell carcinoma of the skin. In contrast, the level of evidence regarding the association of Chlamydia spp. with cancer is low. Mycobacterium tuberculosis is associated with lung cancer, albeit probably not etiopathogenetically.
_
|
Pathogens |
Main associated cancer |
Level of evidence High vs Low* |
|
Salmonella Typhi |
Gallbladder carcinoma Hepatobiliary carcinoma Carcinomas in pancreas, lung and colorectum |
High High Low |
|
Chlamydia species 1.Chlamydia pneumoniae 2. Chlamydia trachomatis 3. Chlamydia psittaci |
Lung carcinoma Ovarian carcinoma Ocular lymphoma |
High Low Low |
|
Mycobacterium tuberculosis |
Lung carcinoma Kaposi’s sarcoma |
Low Low |
|
Schistosoma species 1.Schistosoma haematobium
2. Schistosoma mansoni 3. Schistosoma japonicum |
Bladder carcinoma Cervical carcinoma Colorectal carcinoma Liver carcinoma Colorectal carcinoma |
High Low Low Low Low |
|
Tropheryma whippelii |
Lymphoma Gastric adenocarcinoma |
Low Low |
|
Liver flukes 1.Opisthorchis viverrini 2. Clonorchis sinensis |
Cholangiocarcinoma Cholangiocarcinoma |
High Low |
|
Various causative agents of Chronic Osteomyelitis |
Squamous cell carcinoma of the skin Basal cell carcinoma of the skin Fibrosarcoma Myeloma Angiosarcoma Rhabdomyosarcoma |
High Low Low Low Low Low |
|
Various causative agents of Hidradenitis Suppurativa |
Lymphoma Squamous cell carcinoma of the skin Liver carcinoma |
Low High Low |
*Based on the available published data
_
In conclusion, there is no doubt that specific bacteria species and parasites can cause cancer through different and complex mechanisms. It is of note, however, that although the bacterial species associated with cancer are diverse, they share an important common characteristic: the time-interval between bacterial infection and cancer development, in the majority of cases, is often many years. The paradigms of the tumorigenic action of S. Typhi, as well as the cases of chronic osteomyelitis and hidradenits suppurativa, are quite representative. Additionally, the clinical evidence regarding the association of each pathogen with malignant diseases varies significantly. For some bacterial species (e.g., S. Typhi) this relationship is well established, whereas for other pathogens (e.g., C. psittaci and ocular lymphomas) it is somewhat weak, making further studies necessary.
_____
______
Section-9
Cancer and exercise:
The relationship between physical activity and cancer risk:
Evidence linking higher physical activity to lower cancer risk comes mainly from observational studies, in which individuals report on their physical activity and are followed for years for diagnoses of cancer. Although observational studies cannot prove a causal relationship, when studies in different populations have similar results and when a possible mechanism for a causal relationship exists, this provides evidence of a causal connection.
There is strong evidence that higher levels of physical activity are linked to lower risk of several types of cancer.
- Bladder cancer: In a 2014 meta-analysis of 11 cohort studies and 4 case-control studies, the risk of bladder cancer was 15% lower for individuals with the highest level of recreational or occupational physical activity than in those with the lowest level. A pooled analysis of over 1 million individuals found that leisure-time physical activity was linked to a 13% reduced risk of bladder cancer.
- Breast cancer: Many studies have shown that physically active women have a lower risk of breast cancer than inactive women. In a 2016 meta-analysis that included 38 cohort studies, the most physically active women had a 12–21% lower risk of breast cancer than those who were least physically active. Physical activity has been associated with similar reductions in risk of breast cancer among both premenopausal and postmenopausal women. Women who increase their physical activity after menopause may also have a lower risk of breast cancer than women who do not.
- Colon cancer: In a 2016 meta-analysis of 126 studies, individuals who engaged in the highest level of physical activity had a 19% lower risk of colon cancer than those who were the least physically active.
- Endometrial cancer: Several meta-analyses and cohort studies have examined the relationship between physical activity and the risk of endometrial cancer (cancer of the lining of the uterus). In a meta-analysis of 33 studies, highly physically active women had a 20% lower risk of endometrial cancer than women with low levels of physical activity. There is some evidence that the association is indirect, in that physical activity would have to reduce obesity for the benefits to be observed. Obesity is a strong risk factor for endometrial cancer.
- Esophageal cancer: A 2014 meta-analysis of nine cohort and 15 case–control studies found that the individuals who were most physically active had a 21% lower risk of esophageal adenocarcinoma than those who were least physically active.
- Kidney (renal cell) cancer: In a 2013 meta-analysis of 11 cohort studies and 8 case–control studies, individuals who were the most physically active had a 12% lower risk of renal cancer than those who were the least active. A pooled analysis of over 1 million individuals found that leisure-time physical activity was linked to a 23% reduced risk of kidney cancer.
- Stomach (gastric) cancer: A 2016 meta-analysis of 10 cohort studies and 12 case–control studies reported that individuals who were the most physically active had a 19% lower risk of stomach cancer than those who were least active.
There is some evidence that physical activity is associated with a reduced risk of lung cancer. However, it is possible that differences in smoking, rather than in physical activity, are what explain the association of physical activity with reduced risk of lung cancer. In a 2016 meta-analysis of 25 observational studies, physical activity was associated with reduced risk of lung cancer among former and current smokers but was not associated with risk of lung cancer among never smokers.
For several other cancers, there is more limited evidence of an association. These include certain cancers of the blood, as well as cancers of the pancreas, prostate, ovaries, thyroid, liver, and rectum.
_
Data from more than 35 observational studies suggest that exercise confers a protective effect against cancer-specific mortality, cancer recurrence and all-cause mortality in some cancers. These data show that patients who exercise regularly have a 21 – 35% lower relative risk of cancer recurrence, a 28 – 44% reduced relative risk of cancer-specific mortality and a 25 – 48% decreased relative risk of all-cause mortality when compared with patients who do little or no exercise (Figure below). Most evaluations to date have involved patients with breast, colorectal or prostate cancer, so it is unclear whether exercise is associated with improved disease outcomes in patients diagnosed with other cancer types.
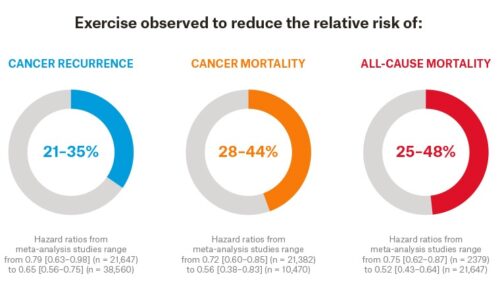
Figure above shows that exercise confers a protective effect against cancer recurrence, cancer-specific mortality and all-cause mortality in some cancers (data arises from studies involving predominately patients with breast, colorectal and prostate cancer).
_
Given the nature of epidemiological investigations, this research is limited by the inability to infer direct causality between exercise behaviour and cancer outcomes. Thus, it is possible that observations of the protective effect of exercise may reflect reverse causality rather than a physiological effect. That is, better outcomes may be reported for more active patients because they are less encumbered by advanced or aggressive disease and/or severe symptomology rather than as a result of exercise-induced adaptations that slow cancer progression. However, the protective effect of exercise was observed in multivariable adjusted analyses that accounted for a range of clinically relevant covariates associated with cancer progression such as cancer stage, treatments, smoking status, body mass index and comorbidities. Furthermore, a range of physiological adaptations that modulate tumour progression have been hypothesised to contribute to these findings. These include favourable exercise-related changes to blood perfusion and tumour vascularisation, immune function, inflammation, oxidative stress and metabolism. Preclinical investigations involving mouse models have shown that exercise in mice undergoing treatment for breast or prostate cancer led to significantly reduced tumour growth and less metastatic disease as well as improved chemotherapeutic efficacy.
_
Various mechanisms underpin the beneficial impact of exercise in cancer:
Exercise provides such widespread benefits to people with cancer because it is a therapy that improves the structure and function of most of the body’s systems simultaneously. In fact, there is no medication or treatment that can positively influence as many body systems as exercise can. As such, a range of potential factors may contribute to the therapeutic effect of exercise for patients with cancer.
Exercise prevents and/or mitigates the negative impact of cancer and its treatment on the musculoskeletal and cardiovascular systems that contribute to physical deconditioning, cancer-related fatigue and other symptoms. By enhancing functional status and reducing treatment-related side effects, exercise may allow patients to tolerate greater dosages of cancer treatment, potentially improving the treatment response. Regular exercise contributes to the resolution of immune homeostasis through modulating the systemic milieu of metabolic (e.g. insulin, glucose, insulin-like growth factor 1), inflammatory (e.g. C-reactive protein, interleukin 6) and sex steroid (e.g. oestrogen) markers. Exercise may enhance the effectiveness of anti-cancer treatments by normalising the tumour microenvironment and potentially increasing the transport of systemic therapies to cancer cells. Exercise may also enhance outcomes through the potential of epigenetic modifications that are concordant with health-enhancing phenotypic adaptations. exercise helps to prevent obesity, which is a risk factor for many cancers. Exercise oncology studies have demonstrated that exercise modulates both the tumor microenvironment and the robustness of the anti-tumor immune response. Emerging evidence from pre-clinical studies indicate that physical activity / exercise paradigms regulate intratumoral vascular maturity and perfusion, hypoxia, and metabolism and augments the anti-tumor immune response. Furthermore, regular exercise is an established prophylactic measure that reduces the risk of developing comorbid conditions such as heart disease, hypertension, diabetes and osteoporosis. Further research is required to elucidate the precise mechanisms underlying the therapeutic effect of exercise in cancer.
_____
The relationship between being sedentary and the risk of cancer:
Although there are fewer studies of sedentary behavior and cancer risk than of physical activity and cancer risk, sedentary behavior—sitting, reclining, or lying down for extended periods of time (other than sleeping)—is a risk factor for developing many chronic conditions and premature death. It may also be associated with increased risk for certain cancers.
______
______
Section-10
Cancer and stress:
Although chronic stress can lead to many health problems, whether it is linked to cancer is not clear. Studies conducted to date have had varying results. For example,
- One case-control study among Canadian men found an association between workplace stress and the risk of prostate cancer, whereas a similar study did not find such an association.
- A prospective study among more than 100,000 UK women reported no association between the risk of breast cancer and perceived stress levels or adverse life events in the preceding 5 years.
- A 15-year prospective study of Australian women at increased risk of familial breast cancer found no association between acute and chronic stressors, social support, optimism, or other emotional characteristics and the risk of breast cancer.
- In a 2008 meta-analysis of 142 prospective studies among people in Asia, Australasia, Europe, and America, stress was associated with a higher incidence of lung cancer.
- A 2019 meta-analysis of nine observational studies in Europe and North America also found an association between work stress and risk of lung, colorectal, and esophageal cancers.
- A meta-analysis of 12 cohort studies in Europe found no link between work stress and the risk of lung, colorectal, breast, or prostate cancers.
Even when stress appears to be linked to cancer risk, the relationship could be indirect. For example, people under chronic stress may develop certain unhealthy behaviors, such as smoking, overeating, becoming less active, or drinking alcohol, that are themselves associated with increased risks of some cancers.
_____
Work stress and risk of cancer: meta-analysis of 5700 incident cancer events in 116000 European men and women, a 2013 study:
A harmonised measure of work stress, high job strain, was not associated with overall risk of cancer (hazard ratio 0.97, 95% confidence interval 0.90 to 1.04) in the multivariable adjusted analyses. Similarly, no association was observed between job strain and the risk of colorectal (1.16, 0.90 to 1.48), lung (1.17, 0.88 to 1.54), breast (0.97, 0.82 to 1.14), or prostate (0.86, 0.68 to 1.09) cancers. There was no clear evidence for an association between the categories of job strain and the risk of cancer. These findings suggest that work related stress, measured and defined as job strain, at baseline is unlikely to be an important risk factor for colorectal, lung, breast, or prostate cancers.
_____
Study suggests a link between Stress and Cancer:
Stress hormones can alter the behavior of some neutrophils, potentially causing dormant cancer cells to reawaken, a study suggests. In experiments in mice, a stress hormone triggered a chain reaction in immune cells that prompted dormant cancer cells to wake up and form tumors again.
Some cancer treatments can push surviving cancer cells into hibernation. These dormant cells either stop growing or grow very slowly. Because there are so few of them, they’re impossible to find with standard tests, And they don’t usually cause any issues—unless they start growing again. Could immune cells wake up dormant cancer cells? To find out, researchers created dormant cancer cells in the laboratory by genetically engineering lung cancer cells, or by treating lung, ovarian, and breast cancer cells with a common chemotherapy drug. Both kinds of dormant cancer cells survived but didn’t grow. In lab dishes, dormant cells didn’t grow when mixed with B cells or T cells, two kinds of immune cells. But they started growing again when mixed with so-called “pro-tumor” neutrophils. Neutrophils, a kind of white blood cell, are part of the body’s first line of defense against infections. What turns neutrophils rogue if there are no tumors left in a patient’s body? Because some studies have linked chronic stress to cancer progression, the scientists explored the effects of stress on neutrophils. Stress hormones like adrenaline and norepinephrine set off a chain reaction involving neutrophils and dormant cancer cells, the researchers found. In lab dishes, stress hormones caused neutrophils to spit out a protein duo known as S100A8/A9. These proteins made neutrophils produce certain lipids that, in turn, awakened dormant lung cancer cells.
The researchers next explored whether the same mechanism occurred in mice that were stressed from being confined for a few hours a day. Stressed mice had more neutrophils in their lungs and spleens than unstressed mice, the scientists found. The stressed mice also had more S100 proteins in their blood. Dormant lung cancer cells formed tumors in stressed mice but not in unstressed mice.
_____
_____
Section-11
Cancer caused by radiation:
Among the physical agents that give rise to cancer, radiant energy is the main tumour-inducing agent in animals, including humans. Radiation is energy. It can come from unstable atoms that undergo radioactive decay, or it can be produced by machines. Radiation travels from its source in the form of energy waves or energized particles. There are different forms of radiation and they have different properties and effects.
There are two kinds of radiation: non-ionizing radiation and ionizing radiation.
Non-ionizing radiation has enough energy to move atoms in a molecule around or cause them to vibrate, but not enough to remove electrons from atoms. Examples of this kind of radiation are radio waves, visible light and microwaves. Non-ionizing radiation is longer wavelength/lower frequency lower energy, while ionizing radiation is short wavelength/high frequency higher energy.
Ionizing radiation has so much energy it can knock electrons out of atoms, a process known as ionization. Ionizing radiation can affect the atoms in living things, so it poses a health risk by damaging tissue and DNA in genes. Ionizing radiation comes from x-ray machines, cosmic particles from outer space and radioactive elements. Radioactive elements emit ionizing radiation as their atoms undergo radioactive decay. Radioactive decay is the emission of energy in the form of ionizing radiation. The ionizing radiation that is emitted can include alpha particle, beta particle and gamma rays. Radioactive decay occurs in unstable atoms called radionuclides.
_
The most common form of radiation exposure is from the sun. Radiation exposure such as ultraviolet radiation and radioactive material is a risk factor for cancer. Many non-melanoma skin cancers are due to ultraviolet radiation, mostly from sunlight. Sources of ionizing radiation include medical imaging and radon gas. Ionizing radiation is not a particularly strong mutagen. Residential exposure to radon gas, for example, has similar cancer risks as passive smoking. Radiation is a more potent source of cancer when combined with other cancer-causing agents, such as radon plus tobacco smoke. Radiation can cause cancer in most parts of the body, in all animals and at any age. Children are twice as likely to develop radiation-induced leukemia as adults; radiation exposure before birth has ten times the effect. Medical use of ionizing radiation is a small but growing source of radiation-induced cancers. Ionizing radiation may be used to treat other cancers, but this may, in some cases, induce a second form of cancer. It is also used in some kinds of medical imaging.
_
Not all types of electromagnetic radiation are carcinogenic. Low-energy waves on the electromagnetic spectrum including radio waves, microwaves, infrared radiation and visible light are thought not to be because they have insufficient energy to break chemical bonds. Non-ionizing radio frequency radiation from mobile phones, electric power transmission, and other similar sources have been described as a possible carcinogen by the World Health Organization’s International Agency for Research on Cancer. However, studies have not found a consistent link between cell phone radiation and cancer risk. The evidence to date suggests that cell phone use does not cause brain or other kinds of cancer in humans.
Higher-energy radiation, including ultraviolet radiation (present in sunlight), x-rays, and gamma radiation, generally is carcinogenic, if received in sufficient doses. Prolonged exposure to ultraviolet radiation from the sun can lead to melanoma and other skin malignancies. The vast majority of non-invasive cancers are non-melanoma skin cancers caused by non-ionizing ultraviolet radiation. Clear evidence establishes ultraviolet radiation, especially the non-ionizing medium wave UVB, as the cause of most non-melanoma skin cancers, which are the most common forms of cancer in the world.
______
Following are the types of non-ionising radiation:
Ultraviolet radiation
Visible light
Infrared
Microwave
Radio waves
Very Low Frequency (VLF)
Extremely Low Frequency (ELF)
Thermal radiation
Black-body radiation
_
Ultraviolet radiation:
Ultraviolet (UV) rays in sunlight give rise to basal-cell carcinoma, squamous-cell carcinoma, and malignant melanoma of the skin. The carcinogenic activity of UV radiation is attributable to the formation of pyrimidine dimers in DNA. Pyrimidine dimers are structures that form between two of the four nucleotide bases that make up DNA—the nucleotides cytosine and thymine, which are members of the chemical family called pyrimidines. If a pyrimidine dimer in a growth regulatory gene is not immediately repaired, it can contribute to tumour development. The risk of developing UV-induced cancer depends on the type of UV rays to which one is exposed (UV-B rays are thought to be the most-dangerous), the intensity of the exposure, and the quantity of protection that the skin cells are afforded by the natural pigment melanin. Fair-skinned persons exposed to the sun have the highest incidence of melanoma because they have the least amount of protective melanin. It is likely that UV radiation is a complete carcinogen—that is, it can initiate and promote tumour growth—just as some chemicals are.
_
Sunlight Spectrum and Wavelengths Responsible for Skin Cancer:
The ultraviolet portion of the solar spectrum is undoubtedly the major factor in skin cancer. Ultraviolet radiation (UVR) is divided into three wavelength ranges on the basis of differences in photochemistry and biologic importance. UVA (320–400 nm) is photocarcinogenic and involved in photoaging but is weakly absorbed in DNA and protein. The relevant chromophores may therefore involve targets that result in production of active oxygen and free radicals, which secondarily cause damage to DNA. UVB (290–320 nm) overlaps the upper end of the DNA and protein absorption spectra and is the range mainly responsible for skin cancer through direct photochemical damage to DNA. UVC (240–290 nm) is not present in ambient sunlight but is readily produced by low pressure mercury sterilizing lamps. The peak wavelength of mercury excitation (254 nm) coincides with the peak of DNA absorption (260 nm), and this wavelength has been of major importance in experimental studies. Absorption of UVR by stratospheric ozone greatly attenuates these wavelengths so that negligible light shorter than 300 nm reaches the earth’s surface. Hence, although UVA and UVB light constitute a minute portion of the emitted solar wavelengths (0.0000001%), they are primarily responsible for the sun’s pathologic effects. Physical shielding of the critical cells of the skin is achieved by the melanin pigment and keratin layers; intracellular defenses depend upon repair of DNA damage, antioxidant enzymes (superoxide dismutase, glutathione reductase, etc.), endogenous free radical quenchers, and inducible detoxifying enzymes and biochemical systems. Melanin itself may play two opposite roles: not only shielding cells from direct UV damage, but indirectly producing damaging free radicals through UV-stimulated redox reactions.
_____
Types of ionizing radiation:
Alpha particles
Beta particles
Gamma rays
X-rays
Ionizing radiation, both electromagnetic and particulate, is a powerful carcinogen, although several years can elapse between exposure and the appearance of a tumour. The contribution of radiation to the total number of human cancers is probably small compared with the impact of chemicals, but the long latency of radiation-induced tumours and the cumulative effect of repeated small doses make precise calculation of its significance difficult.
The carcinogenic effects of ionizing radiation first became apparent at the turn of the 20th century with reports of skin cancer in scientists and physicians who pioneered the use of X-rays and radium. Some medical practices that used X-rays as therapeutic agents were abandoned because of the high increase in the risk of leukemia. The atomic explosions in Japan at Hiroshima and Nagasaki in 1945 provided dramatic examples of radiation carcinogenesis: after an average latency period of seven years, there was a marked increase in leukemia, followed by an increase in solid tumours of the breast, lung, and thyroid. A similar increase in the same types of tumours was observed in areas exposed to high levels of radiation after the Chernobyl disaster in Ukraine in 1986. Electromagnetic radiation is also responsible for cases of lung cancer in uranium miners in central Europe and the Rocky Mountains of North America. Cancers associated with high dose exposure include leukemia, breast, bladder, colon, liver, lung, esophagus, ovarian, multiple myeloma, and stomach cancers.
_

_______
_______
Epidemiology:
Cancer is a stochastic effect of radiation, meaning that it only has a probability of occurrence, as opposed to deterministic effects which always happen over a certain dose threshold. The consensus of the nuclear industry, nuclear regulators, and governments, is that the incidence of cancers due to ionizing radiation can be modelled as increasing linearly with effective radiation dose at a rate of 5.5% per sievert. There is general agreement that the risk is much higher for infants and fetuses than adults, higher for the middle-aged than for seniors, and higher for women than for men, though there is no quantitative consensus about this. Radiation can cause cancer in most parts of the body, in all animals, and at any age, although radiation-induced solid tumors usually take 10–15 years, and can take up to 40 years, to become clinically manifest, and radiation-induced leukemia typically require 2–9 years to appear. Some people, such as those with nevoid basal cell carcinoma syndrome or retinoblastoma, are more susceptible than average to developing cancer from radiation exposure. Children and adolescents are twice as likely to develop radiation-induced leukemia as adults; radiation exposure before birth has ten times the effect. Radiation exposure can cause cancer in any living tissue, but high-dose whole-body external exposure is most closely associated with leukemia, reflecting the high radiosensitivity of bone marrow. Internal exposures tend to cause cancer in the organs where the radioactive material concentrates, so that radon predominantly causes lung cancer, iodine-131 for thyroid cancer is most likely to cause leukemia.
_
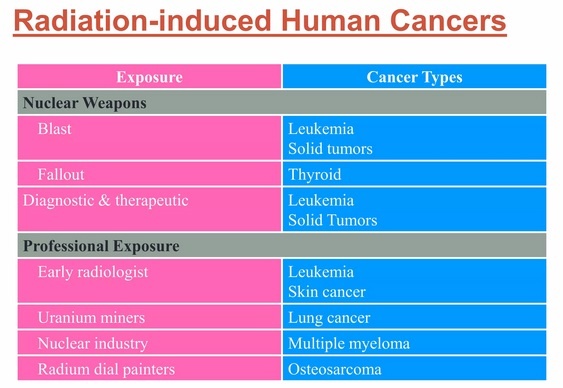
_
Although radiation may cause cancer at high doses and high dose rates, public health data do not absolutely establish the occurrence of cancer following exposure to low doses and dose rates — below about 10,000 mrem (100 mSv). Studies of occupational workers who are chronically exposed to low levels of radiation above normal background have shown no adverse biological effects. Even so, the radiation protection community conservatively assumes that any amount of radiation may pose some risk for causing cancer and hereditary effect, and that the risk is higher for higher radiation exposures. A linear no-threshold (LNT) dose-response relationship is used to describe the relationship between radiation dose and the occurrence of cancer. This dose-response model suggests that any increase in dose, no matter how small, results in an incremental increase in risk. The U.S. Nuclear Regulatory Commission (NRC) accepts the LNT hypothesis as a conservative model for estimating radiation risk.
_
Odds of Cancer:
We do know that radiation-induced cancers do not appear until at least 10 years after exposure (for tumors) or 2 years after exposure (for leukemia). This time after exposure to possible cancer formation is called the “latent period.” The risk of cancer after exposure can extend beyond this latent period for the rest of your life for tumors or about 30 years for leukemia. With cancer, it’s about the risk or odds (probability) of getting cancer. Keep in mind that our everyday, ordinary risk is about 42 percent or odds of 1 in 2.4 without radiation exposure over a lifetime.
|
|
Number of Cancers That Occur over a Lifetime in a Population of One Million People |
Odds of Cancer (natural occurrence) |
|
Cancer Baseline |
420,000* |
1 in 2.4 |
|
POSSIBLE INCREASE IN CANCER CAUSED BY EXPOSURE TO RADIATION ABOVE THE NATURALLY OCCURRING BACKGROUND |
||
|
Dose |
# Possible Cancers if One Million People Receive That Dose |
Combined Odds of Cancer (natural occurrence + the additional risk if you receive the dose in column 1**) |
|
10 mSv |
421,700 |
1 in 2.4 |
|
100 mSv |
437,000 |
1 in 2.3 |
|
1,000 mSv |
590,000 |
1 in 1.7 |
|
10,000 mSv |
A person would die before cancer could occur |
|
|
*Average male plus female lifetime incidence **Adapted from ICRP 2007, Appendix A, Table A14 (17 percent/Sv [or 17 percent/1,000 mSv] incidence) |
||
______
Dose limits of radiation exposure:
Dose limits help ensure that no person is exposed to an excessive amount of radiation in normal, planned situations. They are the strongest form of restriction on dose to an individual. Exceeding a dose limit is contrary to regulations in most countries.
Dose Limits Recommended by ICRP:
|
Type of Dose Limit |
Limit on Dose from Occupational Exposure |
Limit on Dose from Public Exposure |
|
Effective Dose |
20 mSv per year, averaged over defined periods of 5 years, with no single year exceeding 50 mSv |
1 mSv in a year |
|
Equivalent Dose to the Lens of the Eye |
20 mSv per year, averaged over defined periods of 5 years, with no single year exceeding 50 mSv |
15 mSv in a year |
|
Equivalent Dose to the Skin |
500 mSv in a year |
50 mSv in a year |
|
Equivalent Dose to the Hands and Feet |
500 mSv in a year |
– |
Dose limits alone are not enough to ensure adequate protection. They function in combination with the fundamental principles of justification and optimisation. These limits apply only to doses received above the normal local natural background radiation.
Note:
The sievert (Sv) is the International System of Units (SI) derived unit of dose equivalent radiation that takes into account the relative biological effectiveness of different forms of ionizing radiation. One sievert dose equivalent equal to 1 Joule/kilogram that equals 100 Rad that equals 1 Gray for x-rays and gamma rays.
_______
Mechanism:
Cancer is a stochastic effect of radiation, meaning it is an unpredictable event. The probability of occurrence increases with effective radiation dose, but the severity of the cancer is independent of dose. The speed at which cancer advances, the prognosis, the degree of pain, and every other feature of the disease are not functions of the radiation dose to which the person is exposed. This contrasts with the deterministic effects of acute radiation syndrome which increase in severity with dose above a threshold. Cancer starts with a single cell whose operation is disrupted. Normal cell operation is controlled by the chemical structure of DNA molecules. When radiation deposits enough energy in organic tissue to cause ionization, this tends to break molecular bonds, and thus alter the molecular structure of the irradiated molecules. Less energetic radiation, such as visible light, only causes excitation, not ionization, which is usually dissipated as heat with relatively little chemical damage. Ultraviolet light is usually categorized as non-ionizing, but it is actually in an intermediate range that produces some ionization and chemical damage. Hence the carcinogenic mechanism of ultraviolet radiation is similar to that of ionizing radiation.
_
Unlike chemical or physical triggers for cancer, penetrating radiation hits molecules within cells randomly. Molecules broken by radiation can become highly reactive free radicals that cause further chemical damage. Some of this direct and indirect damage will eventually impact chromosomes and epigenetic factors that control the expression of genes. Cellular mechanisms will repair some of this damage, but some repairs will be incorrect and some chromosome abnormalities will turn out to be irreversible. Up to 10% of invasive cancers are related to radiation exposure, including both non-ionizing radiation and ionizing radiation. Unlike chemical or physical triggers for cancer, ionizing radiation hits molecules within cells randomly. If it happens to strike a chromosome, it can break the chromosome, result in an abnormal number of chromosomes, inactivate one or more genes in the part of the chromosome that it hit, delete parts of the DNA sequence, cause chromosome translocations, or cause other types of chromosome abnormalities. Major damage normally results in the cell dying, but smaller damage may leave a stable, partly functional cell that may be capable of proliferating and developing into cancer, especially if tumor suppressor genes were damaged by the radiation. Even if the radiation particle does not strike the DNA directly, it triggers responses from cells that indirectly increase the likelihood of mutations.
_
DNA double-strand breaks (DSBs) are generally accepted to be the most biologically significant lesion by which ionizing radiation causes cancer. In vitro experiments show that ionizing radiation cause DSBs at a rate of 35 DSBs per cell per Gray, and removes a portion of the epigenetic markers of the DNA, which regulate the gene expression. Most of the induced DSBs are repaired within 24h after exposure, however, 25% of the repaired strands are repaired incorrectly and about 20% of fibroblast cells that were exposed to 200mGy died within 4 days after exposure. A portion of the population possess a flawed DNA repair mechanism, and thus suffer a greater insult due to exposure to radiation.
_
Major damage normally results in the cell dying or being unable to reproduce. This effect is responsible for acute radiation syndrome, but these heavily damaged cells cannot become cancerous. Lighter damage may leave a stable, partly functional cell that may be capable of proliferating and eventually developing into cancer, especially if tumor suppressor genes are damaged. The latest research suggests that mutagenic events do not occur immediately after irradiation. Instead, surviving cells appear to have acquired a genomic instability which causes an increased rate of mutations in future generations. The cell will then progress through multiple stages of neoplastic transformation that may culminate into a tumor after years of incubation. The neoplastic transformation can be divided into three major independent stages: morphological changes to the cell, acquisition of cellular immortality (losing normal, life-limiting cell regulatory processes), and adaptations that favor formation of a tumor.
In some cases, a small radiation dose reduces the impact of a subsequent, larger radiation dose. This has been termed an ‘adaptive response’ and is related to hypothetical mechanisms of hormesis.
A latent period of decades may elapse between radiation exposure and the detection of cancer. Those cancers that may develop as a result of radiation exposure are indistinguishable from those that occur naturally or as a result of exposure to other carcinogens. Furthermore, National Cancer Institute literature indicates that chemical and physical hazards and lifestyle factors, such as smoking, alcohol consumption, and diet, significantly contribute to many of these same diseases. Evidence from uranium miners suggests that smoking may have a multiplicative, rather than additive, interaction with radiation. Evaluations of radiation’s contribution to cancer incidence can only be done through large epidemiological studies with thorough data about all other confounding risk factors.
_____
_____
Does radiation therapy cause cancer?
It has long been known that radiation therapy can slightly raise the risk of getting another cancer. It’s one of the possible side effects of treatment that doctors have to think about when they weigh the benefits and risks of each treatment. For the most part, the risk of a second cancer from these treatments is small and is outweighed by the benefit of treating the cancer, but the risk is not zero. This is one of the many reasons each case is different and each person must be part of deciding which kind of treatment is right for them. The risk is different depending on where the radiation treatment will be in the body. If your cancer care team recommends radiation treatment, it’s because they believe that the benefits you’ll get from it will outweigh the possible side effects. Still, this is your decision to make. Knowing as much as you can about the possible benefits and risks can help you be sure that radiation therapy is best for you.
______
______
Is the radiation from CT harmful?
Some people may be concerned about the amount of radiation they receive during CT. CT imaging involves the use of x-rays, which are a form of ionizing radiation. Exposure to ionizing radiation is known to increase the risk of cancer. Standard x-ray procedures, such as routine chest x-rays and mammography, use relatively low levels of ionizing radiation. The radiation exposure from CT is higher than that from standard x-ray procedures, but the increase in cancer risk from one CT scan is still small. Not having the procedure can be much more risky than having it, especially if CT is being used to diagnose cancer or another serious condition in someone who has signs or symptoms of disease.
_
It is also important to note that everyone is exposed to some background level of naturally occurring ionizing radiation every day. The average person in the United States receives an estimated effective dose of about 3 millisieverts (mSv) per year from naturally occurring radioactive materials, such as radon and radiation from outer space. By comparison, the radiation exposure from one low-dose CT scan of the chest (1.5 mSv) is comparable to 6 months of natural background radiation, and a regular-dose CT scan of the chest (7 mSv) is comparable to 2 years of natural background radiation. Effective radiation dose of ultra-low-dose chest CT can be as low as 0.14–0.5 mSv. A single chest x-ray exposes the patient to about 0.1 mSv. This is about the same amount of radiation people are exposed to naturally over the course of about 10 days. The effective doses from diagnostic CT procedures are typically estimated to be in the range of 1 to 10 mSv. This range is not much less than the lowest doses of 5 to 20 mSv received by some of the Japanese survivors of the atomic bombs.
_
The widespread use of CT and other procedures that use ionizing radiation to create images of the body has raised concerns that even small increases in cancer risk could lead to large numbers of future cancers. People who have CT procedures as children may be at higher risk because children are more sensitive to radiation and have a longer life expectancy than adults. Women are at a somewhat higher risk than men of developing cancer after receiving the same radiation exposures at the same ages. People considering CT should talk with their doctors about whether the procedure is necessary for them and about its risks and benefits. Some organizations recommend that people keep a record of the imaging examinations they have received in case their doctors don’t have access to all of their health records.
_
What are the risks of CT scans for children?
Radiation exposure from CT scans affects adults and children differently. Children are considerably more sensitive to radiation than adults because of their growing bodies and the rapid pace at which the cells in their bodies divide. In addition, children have a longer life expectancy than adults, providing a larger window of opportunity for radiation-related cancers to develop.
Individuals who have had multiple CT scans before the age of 15 were found to have an increased risk of developing leukemia, brain tumors, and other cancers in the decade following their first scan. However, the lifetime risk of cancer from a single CT scan was small—about one case of cancer for every 10,000 scans performed on children.
In talking with health care providers, three key questions that the parents can ask are: why is the test needed? Will the results change the treatment decisions? Is there an alternative test that doesn’t involve radiation? If the test is clinically justified, then the parents can be reassured that the benefits will outweigh the small long-term risks.
_____
_____
Section-12
Hormones and cancer:
Some hormones play a role in the development of cancer by promoting cell proliferation. Insulin-like growth factors and their binding proteins play a key role in cancer cell growth, differentiation and apoptosis, suggesting possible involvement in carcinogenesis. Hormones are important agents in sex-related cancers such as cancer of the breast, endometrium, prostate, ovary, and testis, and also of thyroid cancer and bone cancer. For example, the daughters of women who have breast cancer have significantly higher levels of estrogen and progesterone than the daughters of women without breast cancer. These higher hormone levels may explain why these women have higher risk of breast cancer, even in the absence of a breast-cancer gene. Similarly, men of African ancestry have significantly higher levels of testosterone than men of European ancestry, and have a correspondingly much higher level of prostate cancer. Men of Asian ancestry, with the lowest levels of testosterone-activating androstanediol glucuronide, have the lowest levels of prostate cancer. Other factors are also relevant: obese people have higher levels of some hormones associated with cancer and a higher rate of those cancers. Women who take hormone replacement therapy have a higher risk of developing cancers associated with those hormones. On the other hand, people who exercise far more than average have lower levels of these hormones, and lower risk of cancer. Osteosarcoma may be promoted by growth hormones.
_
Some treatments and prevention approaches leverage this cause by artificially reducing hormone levels, and thus discouraging hormone-sensitive cancers. Because steroid hormones are powerful drivers of gene expression in certain cancer cells, changing the levels or activity of certain hormones can cause certain cancers to cease growing or even undergo cell death. Perhaps the most familiar example of hormonal therapy in oncology is the use of the selective estrogen-receptor modulator tamoxifen for the treatment of breast cancer. Another class of hormonal agents, aromatase inhibitors, now have an expanding role in the treatment of breast cancer.
_
Substantial and convincing bodies of experimental, clinical, and epidemiologic evidence indicate that hormones play a major role in the etiology of several human cancers. The concept that hormones can increase the incidence of neoplasia was first proposed by Bittner, on the basis of experimental studies of estrogens and mammary cancer in mice. This theory has been refined into epidemiologic hypotheses related to cancers of the breast, endometrium, prostate, ovary, thyroid, bone, and testis. The underlying mechanism proposed for all of these cancers is that neoplasia is the consequence of prolonged hormonal stimulation of the particular target organ, the normal growth and function of which is controlled by one or more steroid or polypeptide hormones. Evidence is mounting to show that the amount of hormone to which a tissue is effectively exposed is under strong genetic control. Therefore, in addition to external factors such as diet or exogenous hormone use which may modify hormone profiles, polymorphisms in genes encoding proteins involved in steroid-hormone biosynthesis, metabolism or extra- and intracellular transport, and DNA binding are important determinants of individual cancer risk.
_
The major carcinogenic consequence of this hormonal exposure at the end organ is cellular proliferation. The emergence of a malignant phenotype depends on a series of somatic mutations that occur during cell division but the entire sequence of genes involved in progression from normal cell to a particular malignant phenotype are not known (Figure below). Candidate genes include those in the endocrine pathway as well as DNA repair genes, tumor suppressor genes, and oncogenes. Germline mutations have been described in two such tumor suppressor genes, BRCA1 and BRCA2, that have been associated with susceptibility to breast cancer in certain kindreds. Germline mutations in TP53 are also associated in certain kindreds with an increased risk of breast cancer. However, mutations in these genes do not appear to be involved in the majority of sporadic breast cancer. The HER2/neu oncogene is overexpressed in advanced breast cancer, is associated with poor prognosis, and probably represents one critical event in the latter part of breast cancer progression.
_
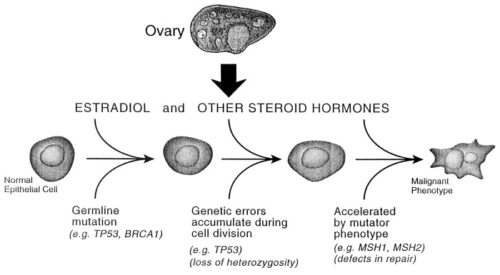
Figure above shows that Estradiol and, to a lesser degree, other steroid hormones (e.g., progesterone) drive breast cell proliferation which facilitates mutation or enhances fixation of mutations or facilitates expression of genetic errors by loss of heterozygosity by defects in DNA repair. Germline mutations in relevant tumor-suppressor genes accelerate the transformation to the malignant phenotype.
_
Neoplasia of hormone-responsive tissues currently accounts for more than 35% of all newly diagnosed male and more than 40% of all newly diagnosed female cancers in the United States. Given that endogenous hormones apparently affect the risk of these cancers and their overall frequency, concern exists about the effects on cancer risk if the same or closely related hormones are administered for therapeutic purposes (e.g., as contraceptives, as hormone replacement therapy, or for the prevention of miscarriage).
_
-1. Breast Cancer:
Available evidence regarding the hormonal etiology of breast cancer is most consistent with the hypothesis that estrogen is the primary stimulant for breast cell proliferation. The simultaneous presence of progesterone probably further increases the rate of proliferation. This latter conclusion is based largely on the fact that breast mitotic activity peaks during the luteal phase of the menstrual cycle.
The most consistently documented, hormonally related risk factors for breast cancer are early age at menarche, late age at menopause, late age at first full-term pregnancy, and weight. The age incidence curve for breast cancer emphasizes the importance of ovulation in determining risk. The initial cases occur during early adulthood, and the rate of increase in incidence then rises sharply with age until the time of menopause, when it slows dramatically. The rate of increase in the postmenopausal period is approximately only one-sixth the rate of increase in the premenopausal period. This age incidence curve appears, then, to be shaped in a major way by the effects of ovarian activity. Therefore, it is critical to understand the determinants, both genetic and environmental, of the onset, regularity, and cessation of ovulation in order to continue to develop effective prevention modalities for breast cancer.
_
Once a breast biopsy is determined to be invasive by a pathologist under the microscope, they will automatically run at least three more tests on the same tissue to determine what “receptors” are present. Receptors are tiny proteins on the surface of the cells that act like “light switches” that can turn cancer cell growth “on” or “off.” The Estrogen receptor (ER), Progesterone receptor (PR), and HER2 receptor results are incredibly important for you to know and understand. These are key determinants as to whether you will or will not benefit from hormonal therapy (pills) or chemotherapy.
When estrogen in your body interacts with an “Estrogen Receptor Positive” cancer, it tends to flip the ER switch of the cancer cells to the “on” or “grow” position. The same can be said for the “Progesterone Receptor.” The HER2 receptor is a little different in that it is not one that interacts with a hormone floating in your body. If the HER2 receptor is found to be “positive” (HER2-Positive) these cancer cells have too many of these types of “on” switches and are always pushing the cancer cells to grow.
Estrogen Receptor Positive (ER+) tumors are always treated with hormonal therapy. Usually, these types of medications (pills) are taken for a total of 5 to 10 years. It is still possible that one may need chemotherapy in addition to hormonal therapy. If you are Progesterone Receptor Positive (PR+) then you will likely need hormonal therapy, even if you are ER-. The Estrogen Receptor plays a much more important role in cancer care than the Progesterone Receptor. Estrogen Receptor Negative (ER-) tumors do not respond to anti-estrogen oral medications that are essential for treating estrogen receptor positive (ER+) tumors. Quite simply, patients with ER-negative tumors will benefit from chemotherapy if they are healthy enough to tolerate it. ER-negative tumors are more aggressive cancers but respond very well to chemotherapy. “Triple Negative” (ER-)(PR-)(HER2-) tumors are also fast-growing tumors that are usually treated with a specific chemotherapy regimen. These tumors are not responsive to hormonal therapy at all but are sensitive to chemotherapy.
______
-2. Endometrial Cancer:
Among the hormone-related cancers, etiologically the best understood is endometrial cancer. All the major demographic characteristics of the disease, as well as the major nondemographic risk factors, are explicable on the basis of cumulative exposure of the endometrium to that fraction of estrogen which is unopposed by the modifying influences of progesterone.
Mitotic Activity in the Endometrium:
Key and Pike have summarized the existing data on endometrial mitotic activity during normal menstrual cycles. Mitotic rates are low during days 1 to 4 of the cycle, then increase rapidly and remain stable thereafter until day 19, after which rates drop to essentially zero for the remainder of the cycle. There appears to be a lag period of about 4 days before the full stimulatory effects of unopposed estrogen, or the modifying influence of progesterone on endometrial mitotic activity, are fully apparent.
The cellular basis for the antiestrogenic activity of progestogens on the endometrium is well understood. Progestogens reduce the concentration of estradiol receptors and increase the activity of the 17ß-hydroxysteroid dehydrogenase type II enzyme (17BHSDII) that converts estradiol to estrone, a biologically less potent estrogen due to its lower affinity for cellular estrogen receptors. Luteal phase progesterone causes endometrial cells to differentiate to a secretory state and progestogen withdrawal leads to cyclic sloughing of endometrial tissue. There are several investigators pursuing the relationship between polymorphisms in the relevant candidate gene and susceptibility to endometrial cancer.
On the basis of the concept that frequency of mitotic activity is the primary determinant of endometrial cancer risk and that such activity is controlled by cumulative exposure to unopposed estrogens, one can readily predict the most important risk factors for this disease. Pregnancies and OCs, which expose the endometrium to constant high levels of both estrogen and progestogen, should protect against endometrial cancer development. Estrogen replacement therapy and obesity should increase the risk. All of these predicted effects have been repeatedly well documented in epidemiologic studies.
____
-3. Ovarian Cancer:
The epidemiology of epithelial ovarian cancer, like breast and endometrial cancer, is well studied. However, the hormonal basis for ovarian cancer appears to differ from that of other hormone-induced cancers, in that one of the most important ovarian epithelial cell mitogens is a gonadotropin, follicle stimulating hormone (FSH). The stimulus for cell division in ovarian cancer etiology is not only hormonal but also a secondary effect following ovulation. In this case, it has been proposed that the epithelial cells within the ovary or covering the ovarian surface are the cells of origin of ovarian cancer. These cells replicate during or after each ovulation; thus, any respite from ovulation would be protective against ovarian cancer. This hypothesis is supported by a large body of epidemiologic data, which consistently demonstrates that the risk of developing ovarian cancer decreases with increasing parity and with combined estrogen-progestin oral contraceptives (COCs) use, both of which induce anovulation.
As with other hormone-related cancers, there is little evidence that any external carcinogen (e.g., tobacco, dietary fat) directly affects the risk of ovarian cancer. There is a small percentage of ovarian cancer linked to BRCA1; however, beyond these rare familial cases, the genetic basis of individual susceptibility to ovarian cancer is largely unstudied. As with breast and endometrial cancers, candidate genes include those involved in steroid biosynthesis (e.g., CYP17) and, importantly, the gonadotropin follicle stimulating hormone (FSH) gene. Estradiol and FSH have growth-stimulating effects on ovarian epithelial cells.
As with breast and endometrial cancer, the age-incidence curve for ovarian cancer emphasizes the importance of ovulation in determining risk. The age-incidence curve of ovarian cancer can be brought into line with the familiar linear log-log plot of other non–hormone-dependent epithelial tumors, if ovarian age is considered as starting at menarche and proceeding at a reduced rate (roughly 30% of normal) during periods of anovulation, including the postmenopausal period.
____
-4. Prostate Cancer:
The prostate is an androgen-regulated organ, and there is little disagreement that androgens are the major stimulus for cell division in prostatic epithelium. Thus androgens are strong candidates as major contributors to prostatic carcinogenesis. Nonetheless, there is still little direct evidence to date that androgens cause prostate cancer and insufficient indirect evidence to make a totally convincing case for a causal relationship, in part because there are no easily measurable hormonal events in men as exist in women (e.g., menarche, menopause, reproductive experiences) that can be related directly to an alteration in prostate cancer risk, and use of exogenous androgens in men is relatively uncommon.
The epidemiology of prostate cancer is dominated by three observations; (1) the profound international and racial-ethnic variation in incidence and mortality, historically reported to be as much as 80-fold between the extremes of high risk (African Americans) and low risk (native Japanese and Chinese) populations; (2) the occurrence of occult, subclinical prostate cancer at a relatively comparable prevalence, albeit much higher rate, among these same populations; and (3) the strong relationship between prostate cancer incidence and aging. Prostate cancer almost never occurs before age 50 years, but it is still the most common cancer of American men, in large part because the rate of increase in prostate cancer incidence with aging is greater than for any other cancer.
Some of the indirect evidence for a role of androgens in prostate cancer development has come from comparisons of hormonal patterns in healthy men from racial-ethnic groups at the extremes and in the middle of prostate cancer incidence. In fact, African American men have higher exposure to testosterone, the main biologically potent circulating androgen, than their Caucasian and Asian counterparts, beginning in the in-utero period. African American women have testosterone levels that exceed those of Caucasian women by 50% or more in early pregnancy, an exposure that has been hypothesized to permanently alter the “gonadostat,” the hypothalamic-pituitary-testicular axis, in African American male offspring relative to Caucasians. African American men during young adulthood also have substantially higher circulating testosterone levels than their Caucasian counterparts (approximately 13 to 15% difference at age 20 years). Although this difference appears to dissipate with age, African American men still have slightly higher testosterone levels than Caucasians (≈3% higher) at age 40 years. Asian men, while showing no evidence of low circulating testosterone levels relative to Caucasians or African Americans at any age, have been shown to have substantially reduced levels of androstanediol glucuronide than either of these other two racial-ethnic groups. This hormone is an index of 5-alpha reductase activity, the prostatic enzyme which bioactivates testosterone to dihydrotestosterone, the most biologically potent human androgen.
Several other indirect lines of evidence point to a role of androgens in prostate cancer pathogenesis. Androgens are required for prostate cancer development or progression in most animal models of prostatic adenocarcinoma. Prostate cancer has never been reported to occur in eunuchs or men with constitutional 5-alpha reductase activity, groups with very low androgen activity and highly underdeveloped prostates. Prostate cancers, at least early in their course, are uniformly androgen dependent, and androgen ablation therapy has been the mainstay for treating early metastatic prostate cancer for many decades. More direct evidence for a role of androgens in prostate carcinogenesis comes from a well-designed prospective study, the Physicians Health Study, which demonstrated that healthy men in the highest quartile of circulating testosterone levels have 2.6 times the likelihood of subsequently developing prostate cancer compared with men in the lowest quartile.
____
-5. Vaginal Adenocarcinoma:
The work of Herbst and colleagues describing the association between in utero diethylstilbestrol (DES) exposure and vaginal adenocarcinoma provided the initial suggestion that estrogen might induce anomalous development in utero which would later have neoplastic consequences in the postpubertal period. These neoplasms developed within a limited age range (approximately ages 15 to 29 years); and the relevant exposure nearly always occurred during the first trimester of the index pregnancy. Vaginal adenocarcinomas appear to develop from Müllerian duct remnants that are induced by DES exposure to persist beyond early fetal life. These remain dormant during childhood and are activated at puberty.
______
-6. Testis Cancer:
The age-specific incidence rates of malignant germ cell tumors of the testis peak in early adult life, in a pattern that is similar to that of DES-induced vaginal adenocarcinoma. This correspondence suggests that the etiology of testicular germ cell neoplasms may also involve in utero hormonal exposure. Risk factors for testis cancer include a history of cryptorchidism, Caucasian race, and in utero exogenous estrogen exposure and may include maternal pregnancy-related nausea and obesity as well.
Men with cryptorchid testes have been reported to have relative risks of testis cancer ranging from 3 to 14, compared with men with normal testicular descent. A persistently undescended testis is often accompanied by other structural abnormalities. The testis is smaller, and tubule development and spermatogenesis are retarded. Sertoli cell development is delayed and Leydig cells are abnormal. It is not the abdominal location of the undescended testis that increases the risk of cancer in the undescended testis. After descent is achieved by surgical treatment, previously undescended testes retain a higher-than-normal risk of cancer. Furthermore, the contralateral, normally descended testis in patients with unilateral cryptorchidism is reported to have a two-fold increase in risk of cancer.
Normal descent of the testis is under hormonal control. Animal experiments have shown that estrogen treatment of pregnant mice can lead to undescended and hypogenetic testes. Similar abnormalities have been reported in the male offspring of women exposed to DES and to OCs during pregnancy. Thus, it is likely that cryptorchidism is tied to estrogen levels early in pregnancy and represents another, sometimes intermediate, outcome of the pathway leading to germ cell testicular tumors.
Other potential risk factors for testis cancer may also be manifestations of excess free maternal estrogen during the critical gestation period. Excessive nausea during pregnancy of mothers of patients with testis cancer may be associated with an increased risk of testis cancer; the risk is greatest in those whose mothers’ nausea required medical treatment, and the risk is most noticeable in sons who were their mothers’ first live birth. Increased levels of bioavailable estradiol are found in the first trimester of pregnancy in women with hyperemesis gravidarum compared with controls and in the first trimester of a woman’s first compared with her second pregnancy. Since adipose tissue is a source of estrogen, the increasing risk of testis cancer observed with increased weight of the mother prior to the index pregnancy may also reflect an excess of bioavailable estrogen.
Three of four studies of the relationship of exogenous sex steroid exposure in pregnancy and testis cancer have shown higher risk of testis cancer in sons who experienced in utero exposure to either DES, OCs, estrogen, or the estrogen-progestin combinations used in pregnancy tests, with reported relative risks ranging from 2.8 to 5.3.
The rarity of testis cancer and, correspondingly, cryptorchidism, in African American males may represent a slight variation of this “estrogen excess” hypothesis. The substantially higher plasma testosterone as well as estrogen levels in African American women compared with Caucasian women early in gestation suggest the possibility that both hormones may be important factors in the development of the testis. The absolute excess of testosterone in the early gestation blood of African American women, by providing a “protected” environment for testicular development and descent, is one possible explanation for the subsequent lower incidence of testis cancer in African American male offspring. In rats, estrogen-inhibited testicular descent can be reversed by treatment with androgens.
____
-7. Cervical Cancer:
Numerous studies of the relationship between hormonal contraceptives and cervical neoplasia have been conducted; however, not much attention has been paid to the contribution of other hormonal factors to the etiology of this disease. A small study of cervical cell mitotic activity during the menstrual cycle has shown that the mitotic rate is almost double during the luteal phase of the cycle than in the follicular phase. The mitotic rate of postmenopausal women was only 33% of the rate of premenopausal women. These data would predict that the slope of the age-specific incidence curve for cervical cancer should decrease after menopause as it does for cancers of the breast, ovary, and endometrium. Mortality and incidence data that predate the widespread use of Pap screening suggest that this is true.
The relationship between oral contraceptives (OCs) and cervical cancer risk requires careful evaluation. Sexual factors such as age at first sexual intercourse and number of sexual partners, which are risk factors for cervical cancer, may also be associated with OC use. Furthermore, OC use is positively associated with the frequency of cervical Pap screening. This positive association would be expected to produce a positive association of cervical carcinoma in situ (CIS) with OC use, whether or not a true etiologic association exists. It would also be expected to downwardly bias any association of OC use with invasive cervical cancer, by detecting tumors at a premalignant or in situ stage. Few studies have adjusted for Pap screening history and sexual history in evaluating the relationship between OC use and cervical cancer risk. Recent case-control studies have examined the issue of a possible spurious association between OC use and cervical CIS but have not produced consistent results.
Analytic studies involving OC use and invasive cervical cancer have focused almost entirely on squamous cell malignancies or have not provided analyses by cell type. Results for studies which included primarily squamous cell tumors provide some evidence that OC use increases the risk of invasive cervical cancer. For example, in a case-control study conducted by Brinton and associates, the relative risk for OC users was 1.5 times that of nonusers, after adjustment for possible confounding by sexual activity risk factors and for Pap screening history. In this study, long-term OC users (5 or more years) had an approximately two-fold higher risk than nonusers. Overall, women who had used OCs with a high estrogen content had the highest risk. Another case-control study found OC use to be a strong predictor of risk of invasive squamous cell cervical cancer, but only if use of OCs began before 18 years of age.
An increasing incidence of adenocarcinoma of the cervix has been reported among women under 35 years of age in Los Angeles County, California and in areas served by population-based cancer registries participating in the Surveillance, Epidemiology, and End Results (SEER) Program. In these areas, incidence of the same tumor type has remained essentially constant over the same period among older women. Peters and co-workers hypothesized that the use of OCs during the teenage years might account for this trend, as OCs produce morphologic changes in the endocervix, characterized by stromal edema, excessive mucus production, and glandular hyperplasia. The extent of these histologic changes increases with longer continuous use of the contraceptive agents. A population-based case-control study conducted in Los Angeles that was limited to young women (born after 1935) who had been diagnosed with adenocarcinoma of the cervix was recently published. On the basis of personal interviews of 195 patients and 386 age-, race- and neighborhood-of-residence–matched controls, the risk of adenocarcinoma of the cervix was greater among women who had used OCs than among those who had not. The highest risk was observed with OC use that exceeded 12 years.
____
-8. Thyroid Cancer:
The pituitary hormone thyroid stimulating hormone (TSH) is the principal hormone regulating the growth and function of the thyroid gland and thus, excess TSH may be of etiologic importance in the development of thyroid cancer. This hypothesis is supported by the observation that growth of some thyroid cancers depends on TSH secretion, so that suppression of TSH release by administration of thyroxin is often an effective treatment for thyroid carcinomas. Experimental studies provide further support for this hypothesis. Sustained elevation of TSH induces thyroid tumors in rodents. The actual mechanism by which elevated TSH levels have been achieved in these studies appears unimportant as thyroid tumors have been produced by iodine-deficient diets, by blocking thyroid hormone synthesis, by administering TSH directly, and by chemical goitrogens.
In the United States, thyroid cancer is roughly 2.5 times more common in women than in men. Incidence rates in women increase sharply from childhood to age 30 years and then level off, whereas in men, thyroid cancer incidence rates increase gradually over the lifespan. The ratio of female-to-male incidence rates is greatest between the ages of 20 and 35 years, during which women have 4 to 5 times the risk of men. This ratio remains above 3 until menopause when it begins to level off around 1.5. This pattern suggests that sex hormones may play an important role in the development of thyroid cancer. A number of epidemiologic studies have shown a strong association between select reproductive factors and thyroid cancer risk. A history of pregnancy has been associated with elevated risk of thyroid cancer in several case-control studies, and the risk was especially elevated among women with pregnancies terminated by spontaneous or induced abortions. Diffuse enlargement of the thyroid gland occurs during pregnancy as a compensatory response to the increased requirement for thyroid hormone production. This alteration in normal thyroid function occurs primarily during the first trimester and seems to level off by 20 weeks of gestation, suggesting that changes in thyroid cells may occur early in pregnancy. It is possible that some of these cellular changes during the first few weeks of pregnancy may alter thyroid cancer risk, and that these changes may be mitigated by full-term pregnancy, but persist following early termination of a pregnancy.
_____
_____
Section-13
Normal cell vs. Cancer cell:
_
- • Normal cell cycle vs. cancer cell cycle:
_
Transcription and translation in protein synthesis:
The process by which DNA is copied to RNA is called transcription, and that by which RNA is used to produce proteins is called translation. Transcription and translation are the two processes that make up the protein synthesis process.
The pathophysiology of cancer is fundamentally an alteration in cellular activity. It is helpful to briefly review how cellular growth and activity is regulated, starting with the central dogma of cell biology: that DNA (deoxyribonucleic acid) carries the genetic code or blueprint for proteins, which in turn help to regulate cellular activity. The basic unit of DNA is a nucleotide, which is a molecule consisting of a 5-carbon sugar (ribose) with an attached phosphate molecule, and one of four different nucleobases: adenine, cytosine, guanine, and thymine.
Nitrogenous bases form complementary pairs via hydrogen bonding (cytosine binds to guanine, adenine to thymine). Adjacent nucleotides form polynucleotide sequences which ultimately comprise the alpha double helix that is DNA. While DNA is understood to form the basis for chromosomes, it is important to remember that DNA is usually tightly packed by associating with histone proteins. When DNA is either replicated or the genetic code is being read, it is unwound and ‘unzippered’ into sense and antisense strands. One strand of DNA is called the sense strand because when you read it in the right direction it provides the code to make a protein. In two-stranded DNA, the sense strand is bonded to an opposite DNA strand that is called the antisense or noncoding strand. In a cell, antisense DNA serves as the template for producing messenger RNA (mRNA), which directs the synthesis of a protein.
The actual genetic code is carried by the sequence of nucleotides in DNA. Each sequence of nucleotides (eg: ATG) corresponds to a complementary sequence in messenger ribonucleic acid (mRNA). For example, ATG corresponds to ‘UAC’ on mRNA, and ultimately this codes for the amino acid tyrosine, while GGC codes for glycine and GUG for valine. Note that the nucleobase thymine (T) is replaced in mRNA with another nucleobase, uracil (U).
The molecular machinery of the cell utilizes this information to produce small amino acid sequences called peptides and longer sequences called proteins. This is a multi-step process, beginning with the production of single-stranded mRNA within the nucleus, a process referred to as transcription. After mRNA leaves the nucleus, the genetic message is ‘read’ or translated by ribosomes, during which individual amino acids are carried by transfer RNA (tRNA).
The triplet of UAC on the mRNA molecule is referred to as a codon. The corresponding nucleobases on the specific, paired tRNA molecule are referred to as an anticodon, and a tRNA with this anticodon carries a specific amino acid, in this case tyrosine.
The pairing of mRNA codons with tRNA anticodons occurs in the cytoplasm at specific sites on ribosomes. As the codon on mRNA matches with its corresponding tRNA, the mRNA moves through the ribosome, making available the next codon of triplet bases. During this process, called translation, amino acids become covalently linked into a growing polypeptide chain.
Peptides and proteins function as either structural components of the cell or enzymes. These molecules in turn catalyze chemical reactions and regulate cellular growth and differentiation. Both transcription and translation must be tightly regulated for normal cellular functioning. Transcription, for example, is regulated by proteins called transcription factors, which bind to specific nucleotide sequences at the start of the coding sequence of a gene called the promoter. Transcription factors enable the gene to be read by another protein, called RNA polymerase. At the end of the gene is another sequence of nucleobases which constitute a ‘stop’ sequence called the terminator.
______
Cell Division:
During a lifetime, many of the cells that make up the body age and die. These cells must be replaced so that the body can continue functioning optimally. Reasons that cells are lost and must be replaced include the following:
- Sloughing off of epithelial cells such as those lining the skin and intestines. The old, worn out cells on the surface of the tissues are constantly replaced. A special case of this is the monthly replacement of the cells lining the uterus in pre-menopausal women.
- Wound healing requires that cells in the area of the damage multiply to replace those lost. Viral diseases such as hepatitis may also cause damage to organs that then need to replace lost cells.
- Replacement of the cells that make up blood. Red blood cells carry oxygen to tissues. White blood cells such as B and T lymphocytes are part of the body’s immune system and help to ward off infections. Most of these cells have very short lifespans and must be constantly replaced. The precursors of these cells are located in bone marrow. These precursors, or stem cells, must reproduce at a very high rate to maintain adequate amounts of the blood cells.
_
The process by which a cell reproduces to create two identical copies of itself is known as mitosis. The goal of mitosis is the formation of two identical cells from a single parent cell. The cells formed are known as daughter cells. In order for this to happen, the following must occur:
- The genetic material, the DNA in chromosomes, must be faithfully copied. This occurs via a process known as replication.
- The organelles, such as mitochondria, must be distributed so that each daughter cell receives an adequate amount to function.
- The cytoplasm of the cell must be physically separated into two different cells.
Many of the features of cancer cells are due to defects in the genes that control cell division. The cell division process occurs as an orderly progression through five different stages. These five stages are collectively known as the cell cycle.
_______
_______
Normal Cell Cycle:
At a fundamental level, cancer is a pathology affecting cell differentiation and growth. Given this fact, a good place for us to start examining this complex disease is to understand the processes which control cell growth and differentiation. Somatic (‘of the body’) cells replicate and divide by the process of mitosis, in which genetic material is replicated and one parent cell divides into two ‘daughter’ cells. In contrast, when germ cells divide, genetic material is halved, a process known as meiosis. Beyond this basic summary, we will not concern ourselves further with the mechanisms and details of mitosis and meiosis. Instead, we will focus on the period between mitosis, called the cell cycle, as depicted below.
_
The cell cycle is an orderly sequence of events. Cells on the path to cell division proceed through a series of precisely timed and carefully regulated stages. The cell cycle is divided into five phases: G0, G1, S, G2 and M. ‘G’ stands for ‘gap’, ‘S’ represents ‘synthesis’, and ‘M’ stands for ‘mitosis’. During mitosis, two processes occur: karyokinesis and cytokinesis. Karyokinesis is the process during which double-stranded chromosomes separate into separate nuclear envelopes, while cytokinesis refers to the process of cell division. After mitosis is complete, two identical daughter cells are produced, which then enter the G1 phase. During G1, cells grow and differentiate into their final forms, and, later in G1, duplicate much of the cellular contents, with the exception of chromosomes. In the event that conditions do not favor cell growth, cells become arrested in G0, where no further growth or differentiation occurs. Some cells, such as muscle and nerve, do not usually divide after differentiation. Others, such as epithelial cells and some connective tissue cells, are capable of cell divisions.
_
The normal mammalian cell cycle has five phases. Between the stages of cell division lie interphases that last at least 12 hours. These ‘resting’ stages or gap phases are badly named; most are periods of intense protein synthesis. Proteins are essential as they make the dividing cell grow big enough to divide. The interphase occurs four times per cycle – Gaps 0, 1, and 2, and the Synthesis (S) phase.
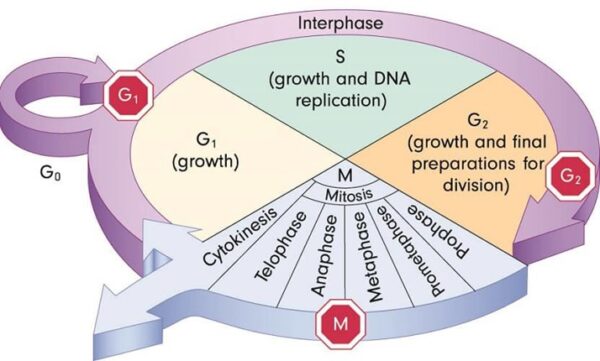
Figure above shows how a cell moves through a series of phases in an orderly manner. During interphase, G1 involves cell growth and protein synthesis, the S phase involves DNA replication and the replication of the centrosome, and G2 involves further growth and protein synthesis. The mitotic phase follows interphase. Mitosis is nuclear division during which duplicated chromosomes are segregated and distributed into daughter nuclei. Usually the cell will divide after mitosis in a process called cytokinesis in which the cytoplasm is divided and two daughter cells are formed. Any part of the cell cycle can stray from the normal pattern.
_
During Gap 0 (G0), the cell is considered out of the cycle. This is the only ‘resting’ interphase. How long a cell remains in this resting period is guided by non-coding DNA. Non-replicating cells do not divide for a temporary period (quiescent cells) or permanently (senescent cells).
During Gap 1 (G1), cells grow larger and produce more proteins. A G1/S checkpoint ensures that the cell is ready to move into the next phase – Synthesis.
At the Synthesis phase (S), the cell must replicate its DNA as it will divide to produce two daughter cells. Each daughter cell must contain a full set of chromosomes to survive.
Gap 2 (G2) signals the end of DNA replication and another period in which more proteins are produced; the cell has another chance to grow in size. Yet another checkpoint ensures the cell is ready to divide. This occurs in the M phase.
At the mitosis (M) phase, the cell stops growing and uses its energy to divide into two identical daughter cells. The M phase involves several steps – prophase, prometaphase, metaphase, anaphase, telophase, and cytokinesis. Mitosis only lasts for an hour or two in mammals and includes a metaphase checkpoint that ensures everything is running smoothly.
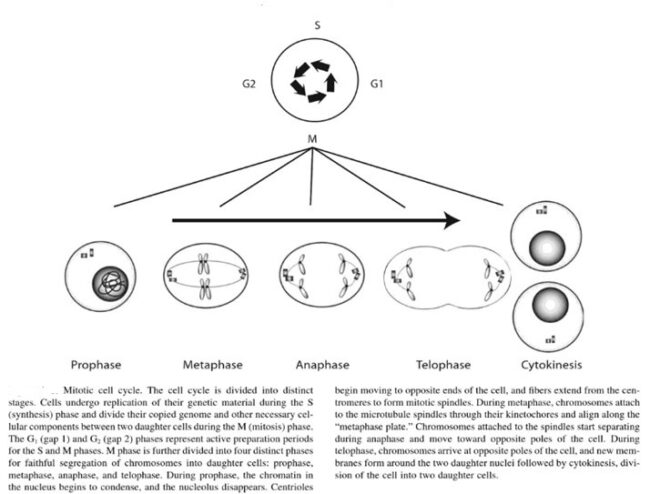
_____
Control of the Cell Cycle:
The length of the cell cycle is highly variable even within the cells of an individual organism. In humans, the frequency of cell turnover ranges from a few hours in early embryonic development to an average of two to five days for epithelial cells, or to an entire human lifetime spent in G0 by specialized cells such as cortical neurons or cardiac muscle cells. There is also variation in the time that a cell spends in each phase of the cell cycle. When fast-dividing mammalian cells are grown in culture (outside the body under optimal growing conditions), the length of the cycle is approximately 24 hours. In rapidly dividing human cells with a 24-hour cell cycle, the G1 phase lasts approximately 11 hours. The timing of events in the cell cycle is controlled by mechanisms that are both internal and external to the cell.
Regulation at Internal Checkpoints:
It is essential that daughter cells be exact duplicates of the parent cell. Mistakes in the duplication or distribution of the chromosomes lead to mutations that may be passed forward to every new cell produced from the abnormal cell. To prevent a compromised cell from continuing to divide, there are internal control mechanisms that operate at three main cell cycle checkpoints at which the cell cycle can be stopped until conditions are favorable. These checkpoints occur near the end of G1, at the G2–M transition, and during metaphase as seen in the figure below.
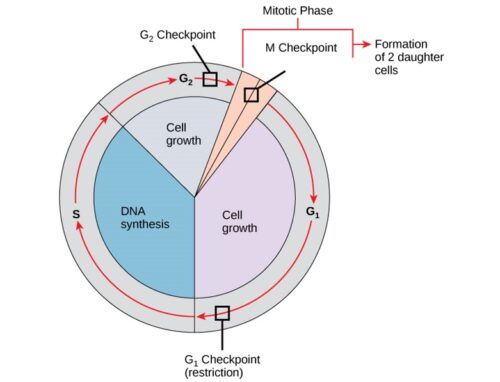
Figure above shows that the cell cycle is controlled at three checkpoints. Integrity of the DNA is assessed at the G1 checkpoint. Proper chromosome duplication is assessed at the G2 checkpoint. Attachment of each kinetochore to a spindle fiber is assessed at the M checkpoint.
The G1 Checkpoint:
The G1 checkpoint determines whether all conditions are favorable for cell division to proceed. The G1 checkpoint, also called the restriction point (R), is the point at which the cell irreversibly commits to the cell-division process. In addition to adequate reserves and cell size, there is a check for damage to the genomic DNA at the G1 checkpoint. A cell that does not meet all the requirements will not be released into the S phase.
The G2 Checkpoint:
The G2 checkpoint bars the entry to the mitotic phase if certain conditions are not met. As in the G1 checkpoint, cell size and protein reserves are assessed. However, the most important role of the G2 checkpoint is to ensure that all of the chromosomes have been replicated and that the replicated DNA is not damaged.
The M Checkpoint:
The M checkpoint occurs near the end of the metaphase stage of mitosis. The M checkpoint is also known as the spindle checkpoint because it determines if all the sister chromatids are correctly attached to the spindle microtubules. Because the separation of the sister chromatids during anaphase is an irreversible step, the cycle will not proceed until the kinetochores of each pair of sister chromatids are firmly anchored to spindle fibers arising from opposite poles of the cell.
_____
Growth factors:
Normal cell division requires constant signals. When the signals are removed, the cells stop dividing.
Cells divide in response to external signals that ‘tell’ them to enter the cell cycle. These signaling molecules bind to their target cells and send signals into the nucleus. The result is that the genes responsible for cell division are turned on and the cell divides. For example, a cut in the skin leads certain blood cells, platelets, to produce a growth factor that causes the skin cells to reproduce and fill the wound. Cell division is a normal process that allows the replacement of dead cells. Progression through the cell cycle, including replication and differentiation, involves a variety of growth-promoting factors; examples of these include epithelial growth factor (EGF), fibroblast growth factor (FGF), and vascular endothelial growth factor (VEGF). Most of these factors bind to extracellular receptors, and activate molecular intermediates including G proteins (e.g.: ras) and kinases which transduce, or transmit, the growth factor signal. The latter typically phosphorylate inhibitory complexes which activate nuclear transcription factors (e.g.: myc, fos, jun). Transcription factors bind to genes which have specific promoter sequences, which begin the processes of transcription. Ultimately, the ‘building blocks’ of life, such as structural proteins and enzymes, are produced as part of the process of growth and differentiation through sequence of growth receptor activation, transduction, and nuclear transcription. A lack of positive external signals causes cells to stop dividing.
Contact inhibition:
Cells are also able to sense their surroundings and respond to changes. For instance, if a cell senses that it is surrounded on all sides by other cells, it will stop dividing. In this way, cells will grow when needed but stop when their goal has been met. To revisit our wound example, the cells fill in the gap left by the wound but then they stop dividing when the gap has been sealed. Cancer cells do not exhibit contact inhibition. They grow even when they are surrounded by other cells causing a mass to form.
Cellular Senescence:
Most cells also seem to have a pre-programmed limit to the number of times that they can divide. Interestingly, the limit seems to be based, in part, on the cell’s ability to maintain the integrity of its DNA. An enzyme, telomerase, is responsible for upkeep of the ends of the chromosomes. In adults, most of our cells don’t utilize telomerase so they eventually die. In cancer cells, telomerase is often active and allows the cells to continue to divide indefinitely.
_______
_______
Cancer Cell Cycle:
The cancer cell cycle uses the same mechanisms as a normal cell, but this process is highly unregulated. A cancer cell cycle would show checkpoint, growth, division, and cell death (apoptosis) anomalies. When errors are found at the checkpoints, a normal cell will usually enter apoptosis and self-destruct. Cancer cell division, however, does not follow the rules of the regulatory checkpoints. With the majority of cancers, gene variants occur in the genes that code for regulatory proteins of the cell cycle. It has been found that the p53 protein malfunctions in more than 60% of cancers. Other regulatory chemicals such as cyclin-dependent kinases bind to proteins called cyclins and, upon doing so, start or stop the cycle. Any gene that interferes with regulatory protein synthesis can lead to continuous cell division and the inability of a damaged cell to self-destruct. As all daughter cells receive the same DNA, a snowball effect occurs. Cancer cells do not grow more quickly than normal cells, but they do not self-destruct and have no limits to how often they divide.
_
Cancer is a collective name for many different diseases caused by a common mechanism: uncontrolled cell division. Despite the redundancy and overlapping levels of cell-cycle control, errors occur. One of the critical processes monitored by the cell-cycle checkpoint surveillance mechanism is the proper replication of DNA during the S phase. Even when all of the cell-cycle controls are fully functional, a small percentage of replication errors (mutations) will be passed on to the daughter cells. If one of these changes to the DNA nucleotide sequence occurs within a gene, a gene mutation results. All cancers begin when a gene mutation gives rise to a faulty protein that participates in the process of cell reproduction. The change in the cell that results from the malformed protein may be minor. Even minor mistakes, however, may allow subsequent mistakes to occur more readily. Over and over, small, uncorrected errors are passed from parent cell to daughter cells and accumulate as each generation of cells produces more non-functional proteins from uncorrected DNA damage. Eventually, the pace of the cell cycle speeds up as the effectiveness of the control and repair mechanisms decreases. Uncontrolled growth of the mutated cells outpaces the growth of normal cells in the area, and a tumor can result.
_
A normal cell becomes a cancer cell when the DNA is damaged in such a way that the cell growth cycle changes. A cancer cell is a non-functioning cell that does not die when it should but continues to divide, passing on its malfunctioning DNA to daughter cells that are also cancer cells. Growth for the sake of growth is the ideology of the cancer cell. The causative factor that turns a normal cell into a cancer cell is the cause of the DNA mutation. There is rarely a single cause. Causes might include a lack of antioxidants in the diet, working without protection in an asbestos factory, chronic inflammation, sunbathing without sunscreen, and inherited mutations.
There are two main groups of gene variants (mutations). These are inherited and non-inherited.
Inherited gene mutations are passed on through generations. Some inherited gene mutations are de novo. The mutation occurs in the sperm of the father or the egg of the mother and the gene variant occurs during fertilization, passing on this DNA to every cell of the growing embryo. If the gene variant exists in proteins that only form later on in life, such as certain hormones during puberty, symptoms may be absent until a later date. Currently, very little can be done to prevent inherited gene variants. Inherited cancer is rare; however, inherited cancer syndromes increase the risk for developing certain cancers. One example is ovarian cancer that often develops in females born in families carrying the gene for hereditary breast and ovarian cancer syndrome. Another example is the increased risk of colon cancer in people from families that carry the hereditary non-polyposis colorectal cancer gene.
Non-inherited gene mutations happen during our lifecycle and only affect individual cells – these can be spread throughout the body or be of the same tissue type. A mutation or gene variant does not always cause cancer but it can cause illness over time. As subsequent generations of cells divide to produce the variant of the original cell, a problem might become evident. The causes of non-inherited gene mutations are many. Inhalation of asbestos dust can damage the DNA of lung cells, as can tobacco smoke, radon gas, and soot. All are carcinogenic – they have the potential to create cancer cells. Obesity increases the risk of developing colorectal, esophageal, kidney, and pancreatic cancer, probably due to a chronic inflammatory state. Infections can also damage DNA and lead to cancer cell development. Remember that only mutated cells that grow uncontrollably are cancer cells.
Ultra-violet radiation and x-rays are sources of radiation that break DNA bonds. When the DNA is repaired incorrectly or radiation exposure continues, there is a higher chance of a cancer cell developing. The larger the area of exposure, the higher the number of potential cancer cells produced. Alcohol is another carcinogen that, over time, affects the DNA of cells in the digestive tract, airways, and liver. This is mainly the effect of the carcinogen acetaldehyde that is produced during the breakdown of alcohol.
As you might expect, the progression of cells through the cell cycle is under a series of tight regulatory controls. As we begin to discuss the biology of cancer, it will become evident that dysregulation or disruption of these controls can lead to alterations in cell growth and differentiation. We can summarize our discussion of regulators of the cell cycle with the notion that it is under the control of a system of “Stop” and “Go” signals.
Two possible scenarios may lead to dysregulated cell growth and, ultimately, carcinogenesis: either a pathway that normally promotes cell growth may become disinhibited, or ‘turned on’, or a process which stops cell growth may become permanently disactivated. In the former scenario, a receptor such as EGF is altered, and activates in the absence of its ligand. Other parts of the pathway, such as receptors, transducers, or transcription factors, may be activated or over-expressed (too much of the protein is made). Conversely, a signaling pathway that stops cell growth may become knocked out (silenced).
In general, two types of genes are implicated in cancer: proto-oncogenes and tumour suppressor genes.
_
Proto-oncogenes:
The genes that code for the positive cell-cycle regulators are called proto-oncogenes. Proto-oncogenes are normal genes that, when mutated, become oncogenes—genes that cause a cell to become cancerous. Consider what might happen to the cell cycle in a cell with a recently acquired oncogene. In most instances, the alteration of the DNA sequence will result in a less functional (or non-functional) protein. The result is detrimental to the cell and will likely prevent the cell from completing the cell cycle; however, the organism is not harmed because the mutation will not be carried forward. If a cell cannot reproduce, the mutation is not propagated and the damage is minimal. Occasionally, however, a gene mutation causes a change that increases the activity of a positive regulator. For example, a mutation that allows Cdk, a protein involved in cell-cycle regulation, to be activated before it should be could push the cell cycle past a checkpoint before all of the required conditions are met. If the resulting daughter cells are too damaged to undertake further cell divisions, the mutation would not be propagated and no harm comes to the organism. However, if the atypical daughter cells are able to divide further, the subsequent generation of cells will likely accumulate even more mutations, some possibly in additional genes that regulate the cell cycle.
The Cdk example is only one of many genes that are considered proto-oncogenes. In addition to the cell-cycle regulatory proteins, any protein that influences the cycle can be altered in such a way as to override cell-cycle checkpoints. Once a proto-oncogene has been altered such that there is an increase in the rate of the cell cycle, it is then called an oncogene.
_
Tumor Suppressor Genes:
Like proto-oncogenes, many of the negative cell-cycle regulatory proteins were discovered in cells that had become cancerous. Tumor suppressor genes are genes that code for the negative regulator proteins, the type of regulator that—when activated—can prevent the cell from undergoing uncontrolled division. The collective function of the best-understood tumor suppressor gene proteins, retinoblastoma protein (RB1), p53, and p21, is to put up a roadblock to cell-cycle progress until certain events are completed. A cell that carries a mutated form of a negative regulator might not be able to halt the cell cycle if there is a problem.
Mutated p53 genes have been identified in more than half of all human tumor cells. This discovery is not surprising in light of the multiple roles that the p53 protein plays at the G1 checkpoint. The p53 protein activates other genes whose products halt the cell cycle (allowing time for DNA repair), activates genes whose products participate in DNA repair, or activates genes that initiate cell death when DNA damage cannot be repaired. A damaged p53 gene can result in the cell behaving as if there are no mutations (Figure below). This allows cells to divide, propagating the mutation in daughter cells and allowing the accumulation of new mutations. In addition, the damaged version of p53 found in cancer cells cannot trigger cell death.
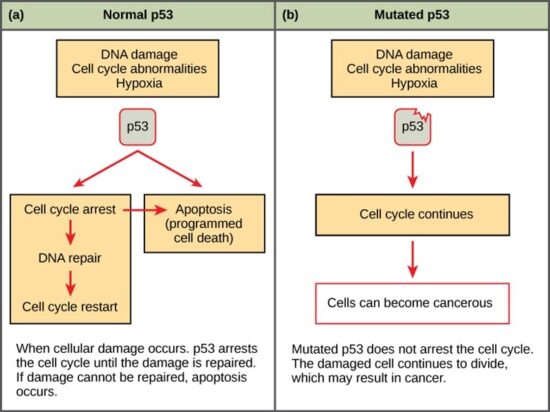
Figure above shows that the role of normal p53 is to monitor DNA and the supply of oxygen (hypoxia is a condition of reduced oxygen supply). If damage is detected, p53 triggers repair mechanisms. If repairs are unsuccessful, p53 signals apoptosis. A cell with an abnormal p53 protein cannot repair damaged DNA and thus cannot signal apoptosis. Cells with abnormal p53 can become cancerous.
_
Cancer is the result of unchecked cell division caused by a breakdown of the mechanisms regulating the cell cycle. The loss of control begins with a change in the DNA sequence of a gene that codes for one of the regulatory molecules. Faulty instructions lead to a protein that does not function as it should. Any disruption of the monitoring system can allow other mistakes to be passed on to the daughter cells. Each successive cell division will give rise to daughter cells with even more accumulated damage. Eventually, all checkpoints become nonfunctional, and rapidly reproducing cells crowd out normal cells, resulting in tumorous growth.
_
Cellular Immortality:
One of the mechanisms behind this involves an enzyme called telomerase. To understand what this enzyme does, we need to first describe the basic structure of a chromosome, which contains tightly-packed DNA, as well as regions which do not contain any genetic information, including centromeres and telomeres. The center of a chromosome is called the centromere, which serves as an attachment point for cytoskeletal proteins during mitosis. At either end of a chromosome are multiple telomeres (‘telos’ being Greek for ‘distant’). Telomeres are nucleoprotein repeats which contain no coding information. Normally, during cell division, telomeres are lost, likely due to mechanical wear. After enough cell divisions, coding regions of the chromosome are also lost, and this is likely one reason why non-cancerous cells can only divide a finite number of times. In certain types of ovarian cancer, an enzyme known as telomerase is over-expressed. Telomerase enzyme replaces telomeres on the ends of chromosomes, and is normally expressed at low levels.
_
Cell division differences between normal cells and cancer cells:
Cell Division in Normal Cells:
- Mechanisms exist to ensure DNA replication occurs correctly and the environmental conditions are favorable for cell division. Replication errors may also be corrected after they occur.
- Normal cells stop dividing when there is genetic damage or conditions are not favorable. Cancer cells continue to divide even when conditions are not appropriate.
- Most cells in the body are not actively dividing, but are carrying out their normal functions.
- Cells divide in response to external signals in the form of protein or steroid growth factors.
- Cells stop dividing for several reasons, including:
-1. A lack of positive external signals
-2. The cell senses that it is surrounded on all sides by other cells-contact dependent (density dependent) inhibition
-3. Most cells seem to have a pre-programmed limit of the number of times they can divide
Cell Division in Cancer Cells:
- Cancer cells can divide without appropriate external signals.
- Cancer cells do no exhibit contact inhibition.
- Cancer cells continue dividing in the presence of genetic damage.
- The uninhibited, continued division of genetically damaged cells can lead to tumor formation.
_______
_______
- • Normal cell properties vs. cancer cell properties:
_
Features of normal cells:
Normal body cells have a number of important features. They can:
- reproduce when and where it’s needed
- stick together in the right place in the body
- self-destruct when they become damaged or too old
- become specialised (mature). This means they have a specific role to perform for example as a muscle cell or red blood cell.
_
The difference between a normal cell and cancer cell is spread across a range of characteristics. Normal cells reproduce according to regulatory signals in the DNA. For example, while nearly all of our cells contain the DNA code that codes for the entire human body, only a select group of information is available to a particular group at a particular time. A white blood cell contains the DNA code to produce insulin, but this code – the insulin-producing gene – is only switched on in the beta cells of the pancreas. We say that the beta-cell expresses the insulin-producing gene. A white blood cell does not express this gene.
While coding DNA produces every protein required to produce the cells, tissues, and organs of the body, non-coding DNA tells cells when and in what quantities to produce them. Gene expression also controls when a cell grows, matures, stops growing, and divides.
In cancer cells, mutations either stop certain proteins from being produced or they stop regulatory genes from working properly. Any gene involved in normal cell growth and division is called a proto-oncogene. Cancer cells do not stop growing or dividing. Instead, one or more of their proto-oncogenes mutates into one or more ‘oncogenes’ – changes to DNA sequences that contribute to cancer development.
Another system that regulates cell growth and death is a group of proteins called growth factors. Cells have receptors for a wide variety of growth factors that tell them when to divide, mature, and die. When the DNA of a cell mutates to become a cancer cell, any of the estimated 25,000 genes in the human genome can be affected. A cell may develop more or fewer receptors for growth factors, be unable to understand the signals, or understand the instructions incorrectly. Cancer cells act differently than normal cells. For example, during the in vitro cell culture condition, normal cells divide only if supplemented with growth factors, but the cancerous cells can divide even in the absence of growth factors. Cancer cells can synthesize the growth factors if they are not available to them. Also, cancer cells do not follow the signals that command them to stop further cell division.
Cells communicate with each other through chemical signals. It is these signals that alert cells when to grow, divide, stop dividing, etc. These signals are also important for preventing groups of cells from encroaching upon the space of other groups of cells. It is well known that in a cancerous cell, one of the first mutations to occur is in its ability to communicate with its surroundings. In absence of this characteristic cells are able to grow rapidly and invade surrounding tissues even though they may be receiving, but not processing appropriately, external signals to halt growth.
Cells in which the DNA is damaged beyond repair are ordered to self-destruct (apoptosis). Normal cells rely on specific proteins that carry out continuous checks on DNA health. These are called tumor-suppressor genes as, by destroying badly-damaged cells, no mutated daughter cells carry on the cancer cell cycle; the original cell is dead and cannot reproduce. Without tumor suppressor proteins, cancer cell tumors can form.
Other genes that are commonly remodeled (mutated) due to pollutants, radiation, unhealthy lifestyles, and any of the countless risk factors for cancer cell development are those that produce cell-adhesion molecules or CAMs. Large groups of normal cells stick together to produce tissues – bone, liver, skin, or muscle, for example. With mutations in CAM-producing genes, cancer cells no longer adhere to their tissue type. This is the basis of metastasis or the movement of cancer cells through the fluids of blood or lymph. These cells can then relocate to other tissues such as the lungs, brain, or liver as metastatic cancer cells.
Another difference between cancer cells and normal cells is cell specialization. When a cell becomes specialized it can carry out its allotted function. A beta cell produces insulin, a muscle cell contracts and relaxes, a white blood cell protects the body from bacteria and viruses. With cancer cells, there is no specialization as these cells never mature. All cancer cells are non-functional.
Another feature of a cancer cell is immortality i.e., they can divide numerous times in contrast to the normal cells. DNA telomere cap maintains the genomic integrity of the normal cells and their progressive shortening occurs during successive cell divisions, but in the case of cancer cells, telomere length is maintained by the telomerase activity which plays a crucial role in cancer cell survival and development. Cancer cells accomplish replicative immortality by activating the silent telomerase encoding gene hTERT (human TERT) that has reverse transcriptase activity which helps to bypass the cancer cell senescence.
The malignant cell is characterized by: acceleration of the cell cycle; genomic alterations; invasive growth; increased cell mobility; chemotaxis; changes in the cellular surface; secretion of lytic factors, etc.
Physical differences between a normal cell and a cancer cell also exist. Cancer cells tend not to be all the same shape and differ from the cell they were intended to be. This can even be seen in the nucleus; a cancer cell nucleus has membrane blebs (bulges).
__
The functional changes of neoplastic cells cause the formation and elimination of active substances, such as: growth factors, hormones, molecules similar to hormones, lytic enzymes, etc. Lytic enzymes (collagenase, cathepsin and plasmogen activator) favor the increased mobility and dissemination of neoplastic cells.
Major alterations occur in energy metabolism, between normal and malignant cells, especially regarding the use of glucose. The energy production with the highest efficiency in cells is performed by oxidation in the tricarboxylic acid cycle (TCA cycle of Krebs cycle), where 36 ATP molecules are produced for each glucose molecule. This metabolism is carried out by oxygen use and represents the main energy production pathway, in the majority of cells. Cancerous cells exhibit anomalies of both glycolysis and the tricarboxylic acid (TCA) cycle. The cancerous cell is particularly characterized by a poor use of oxygen and the massive use of glucose, which is exclusively converted to lactic acid. One glucose molecule generates only 4 ATP. Consequently, malignant cells take from blood a 5–10 fold glucose amount compared to normal cells and they produce a corresponding lactic acid amount that will be recycled and changed back to glucose in the liver. Cancer cells utilize glucose exclusively while normal cells can utilize fat and ketones besides glucose. Tumor cells behave like a metabolic parasite for the organism or they drain its energy.
_
Two additional properties of cancer cells affect their interactions with other tissue components, thereby playing important roles in invasion and metastasis. First, malignant cells generally secrete proteases that digest extracellular matrix components, allowing the cancer cells to invade adjacent normal tissues. Secretion of collagenase, for example, appears to be an important determinant of the ability of carcinomas to digest and penetrate through basal laminae to invade underlying connective tissue. Second, cancer cells secrete growth factors that promote the formation of new blood vessels (angiogenesis). Angiogenesis is needed to support the growth of a tumor beyond the size of few millimeters, at which point new blood vessels are required to supply oxygen and nutrients to the proliferating tumor cells. Such blood vessels are formed in response to growth factors, secreted by the tumor cells, that stimulate proliferation of endothelial cells in the walls of capillaries in surrounding tissue, resulting in the outgrowth of new capillaries into the tumor. The formation of such new blood vessels is important not only in supporting tumor growth, but also in metastasis. The actively growing new capillaries formed in response to angiogenic stimulation are easily penetrated by the tumor cells, providing a ready opportunity for cancer cells to enter the circulatory system and begin the metastatic process.
_
Another general characteristic of most cancer cells is that they fail to differentiate normally. Such defective differentiation is closely coupled to abnormal proliferation, since most fully differentiated cells either cease cell division or divide only rarely. Rather than carrying out their normal differentiation program, cancer cells are usually blocked at an early stage of differentiation, consistent with their continued active proliferation. The leukemia provide a particularly good example of the relationship between defective differentiation and malignancy. All of the different types of blood cells are derived from a common stem cell in the bone marrow. Descendants of these cells then become committed to specific differentiation pathways. Some cells, for example, differentiate to form erythrocytes whereas others differentiate to form lymphocytes, granulocytes, or macrophages. Cells of each of these types undergo several rounds of division as they differentiate, but once they become fully differentiated, cell division ceases. Leukemic cells, in contrast, fail to undergo terminal differentiation. Instead, they become arrested at early stages of maturation at which they retain their capacity for proliferation and continue to reproduce.
______
Scientists have made great strides in the area of cancer research. We now have a better understanding of how cancer cells transition from normal cells as they “gradually become malignant through a progression of alterations” (Cooper, 2000). These changes or mutations are typically genetic in nature and may be facilitated by exposure to tobacco smoke, hormones, certain viruses as well as environmental hazards such as radiation, ultraviolet radiation, and carcinogenic chemicals. A combination of these factors may cause cells to divide uncontrollably and invade adjacent tissues. What are those changes in cell properties that cause cancer cells to differ from normal cells? These differences are outlined in the table below.
|
|
Normal Cells |
Cancer Cells |
|
Growth Factor Proteins |
Stop growing and dividing when they stop producing growth factors, when they have reached their limit, or grown to their maximum. |
May produce their own growth factors that stimulate reproduction and are less dependent on growth factors from other sources. |
|
Density-dependent inhibition |
Stop growing when the cell has reached a finite cell density. |
Continue to divide despite cell density. |
|
Specialization and maturity |
Mature into distinct cell types with specific functions; cell division stops once they become fully differentiated. |
Do not specialize or differentiate; divide quickly before maturing and remain immature and undifferentiated. |
|
Cell-cell interactions (Contact inhibition) |
Respond to signals from adjacent cells telling them they have reached their limit or boundary, causing them to stop growing. |
Do not respond to signals from other cells to stop growing; will grow in a disorganized manner, invading other tissues or migrating over adjacent cells. |
|
Apoptosis (programmed cell death) |
Occurs when a cell is no longer needed, grows old or when DNA damage cannot be repaired. |
Do not repair themselves and do not undergo apoptosis, and thus live longer. |
|
Genes:
|
|
|
|
Cell adhesion molecules |
Secrete cell surface adhesion molecules that make them “stick” to other cells. |
Do not secrete surface adhesion molecules, allowing them to invade other cells and metastasize to distant parts of the body through the bloodstream or lymphatic system. |
|
Morphology (shape and appearance of the cell) |
Uniform in size and shape. |
Vary in size and often have an abnormal shape; nucleus may appear darker due to increased number of DNA strands. |
|
Response to the immune system |
When damaged, lymphocytes remove the cells. |
Evade the immune system by hiding or secreting chemicals that inactivate immune cells. |
|
Angiogenesis (development of new blood vessels) |
Develop new blood vessels to grow and repair damaged tissues. |
Secrete growth factors that promote angiogenesis to support growth. |
|
Chromosome quantity |
Normal quantity |
Abnormal quantity |
|
Telomeres (caps at the end of each strand of DNA that protects the chromosomes) |
When cells divide, telomeres shorten; once they become too short, a cell can’t divide and dies. |
Capable of regenerating telomeres and continue to divide. |
______
______
Cancer cells under microscope:
Cancer cells have distinguishing histological features visible under the microscope. The nucleus is often large and irregular, and the cytoplasm may also display abnormalities.
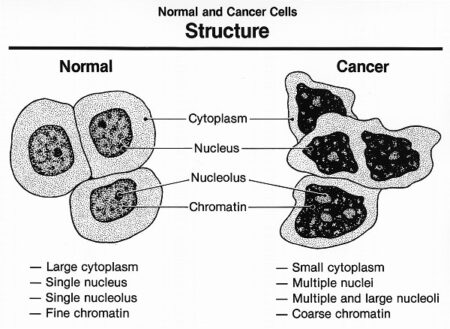
Although there are many forms of cancer, we can generalize about the physical and functional traits of cancerous cells. Physically, cancer cells differ from normal cells in the following ways:
-1. They have larger, irregularly-shaped nuclei
-2. Cell shape is altered or irregular
-3. Cancer cells typically are dedifferentiated and/or express unusual markers; and
-4. They have a high mitotic index
The shape, size, protein composition, and texture of the nucleus are often altered in malignant cells. The nucleus may acquire grooves, folds or indentations, chromatin may aggregate or disperse, and the nucleolus can become enlarged. In normal cells, the nucleus is often round or solid in shape, but in cancer cells the outline is often irregular. In general, cancer cells have larger nuclei than normal cells due to alterations in the DNA that have resulted in the development of cancer. Cancer cells synthesize DNA and RNA far more than normal cells. Tumors and tumor-derived cell lines contain polyploid giant cells with significantly elevated genomic content, often with multiple nuclei. Different combinations of abnormalities are characteristic of different cancer types, to the extent that nuclear appearance can be used as a marker in cancer diagnostics and grading. Mutated structural genes may produce proteins that cause alterations in cell structure. Finally, many malignant tumours, especially ones that are considered ‘high grade’, have a high proportion of cells undergoing mitosis (high mitotic index). This is consistent with a loss of cell cycle controls. Tissues which are typically lost and replaced at a high rate, such as epithelial cells, have a high mitotic index compared to non-dividing cells, such as muscle or nerve. This is one of the contributing factors as to why we see more cancers in rapidly-dividing cells: there is a greater likelihood of an error with each progression through the cell cycle. Another reason for higher cancer rates among epithelial cells is that these are more directly exposed to mutagenic environmental stimuli. However, cancer cells in any tissue have an inappropriately higher mitotic index. In this context, immortality refers to the fact that, unlike normal cells which divide a finite number of times, cancer cells keep dividing more-or-less indefinitely. Cancer cells also usually (though not always) progress rapidly through the cell cycle.
Note:
Mitotic Index (MI) is the ratio between the number of cells in a population undergoing mitosis to the total number of cells in a population.
______
______
Benign vs. malignant tumours:
Examples of benign tumours include moles, nerve ganglia, cysts, uterine fibroid tumours, and endometriosis. The characteristics of benign tumours include:
- inappropriate cell growth
- benign tissues unnecessary for normal functions
- strongly resemble parent cells; retain morphology of tissue of origin
- specific differentiated functions
- joined tightly together
- do not migrate
- small nucleus to cytoplasm ratio
In contrast, malignant tumours are characterized by:
- appearance changed from cells of origin
- size and shape make identification of the origin more difficult
- poorly controlled growth continuous cell division and growth
- growth compares to the parent cell, for example, bone marrow cells divide rapidly, leukemia cells divide rapidly
- large nucleus to cytoplasm ratio
- loss of differentiated functions/appearance
- migrate through blood vessels and tissues
- altered surface proteins
- break away from tumour with only slight pressure
_
Characteristics of benign and malignant tumors are compared in table below:
|
CHARACTERISTICS |
BENIGN |
MALIGNANT |
|
Growth type |
Expansive |
Infiltrating |
|
Growth speed |
Slow (in general) |
Rapid (in general) |
|
Stabilization |
Frequent |
Exceptional |
|
Structure |
Typical |
Atypical (dedifferentiation − anaplasia) |
|
Mitoses |
Rare + Typical |
Numerous + Atypical |
|
Evolution |
Local |
Local + General |
|
Metastasizing |
No |
Yes |
|
Local consequences |
Variable (compressions, …) |
Severe (infiltration, destruction, necrosis, …) |
|
General consequences |
None (exceptions: secretory tumors or at particular sites) |
Constant + severe (in the generalization phase) |
|
Spontaneous evolution |
Usually favorable |
Always fatal |
|
Evolution after removal |
No recurrences |
Common recurrences |
_
The basal membrane is present in benign tumors, while the invasive growth of malignant cells is characterized by fragmentation, reduplication or disappearance of the basal membrane. During the first phases of malignancy, defects are produced with the interruption of the lamina densa. Malignant cells have lytic factors that destroy the basal membrane. The loss of the basal membrane is considered a fundamental criterion of morphological and biological differentiation between benign and malignant tumors. The basal membrane in malignant cells changes its structure or/and ratios between various components, such as: type IV collagen, laminin, heparan sulfate proteoglycan and fibronectin. Neoplastic cells secrete type IV collagenase that destroys type IV collagen, which facilitates metastasizing through the lysis of basal membranes from blood and lymphatic vessels. Thus, malignant cells are disseminated, but they can also leave the vessels and implant in other tissues and organs, with the formation of metastases. In the process of destruction of the basal membrane, a special role is played by laminin and laminin receptors, receptors that are found in the cell membrane and are reorganized during invasive growth.
_____
Cancer as an organ that ignores its niche:
The fundamental cellular defects that create a malignant neoplasm act at the cellular level, and some of these are cell autonomous. However, that is not the entire story. Cancers consist of both malignant cells as well as other cells, blood vessels, extracellular matrix, and signaling and other molecules in the cancer microenvironment. They behave as organs that have lost their specialized function and stopped responding to signals that would limit their growth in tightly regulated normal tissue homeostasis. Most human cancers usually become clinically detectable when a primary mass is approximately 1 cm in diameter—such a mass consists of about 109 cells. Often, patients present with tumors that are approximately 1010 cells. Although it varies by type of cancer and where the primary tumor and metastases are located, a lethal tumor burden is usually about 1012–1013 cells. If all malignant cells were dividing without any cell death at the time of diagnosis, most patients would reach a lethal tumor burden in a very short time. However, human tumors grow by Gompertzian kinetics—this means that not every daughter cell produced by a cell division is actively dividing. In addition, the overall growth rate of a tumor depends on differences between growth rates of different cells within the tumor and rate of cell loss. The growth fraction of a tumor declines with time, largely due to factors in the microenvironment. The growth fraction of the first malignant cell is 100%, and by the time a patient presents for medical care, the growth fraction is estimated to be <10%, although the fraction varies between different types of cancers and even different cancers of the same type in different individuals. This fraction is often similar to the growth fraction of normal bone marrow and normal intestinal epithelium, the most highly proliferative normal tissues in the human body, a fact that may explain the dose-limiting toxicities to these tissues of agents that target dividing cells. The implication of these data is that the tumor is slowing its own growth over time. How does it do this? The tumor cells have multiple genetic lesions that tend to promote proliferation, yet by the time the tumor is clinically detectable, its capacity for proliferation has declined. Better understanding of how a tumor slows its own growth would provide important clues for better cancer treatment. A number of factors, including those in the tumor microenvironment, are known to contribute to the decreased proliferation of tumor cells over time in vivo. Some cells are hypoxemic and have inadequate supply of nutrients and energy. Some have sustained too much genetic stress to complete the cell cycle but have lost the capacity to undergo apoptosis and therefore survive but do not proliferate. However, an important subset is not actively dividing but retains the capacity to divide and can start dividing again under certain conditions such as when the tumor mass is reduced by treatments leading to improved conditions in the tumor microenvironment favorable for cell proliferation. Just as the bone marrow increases its rate of proliferation in response to bone marrow–damaging agents, the tumor also seems to sense when tumor cell numbers have been reduced and can respond by increasing growth rate. However, the critical difference is that the marrow stops growing when it has reached its production goals, whereas tumors do not. The ultimate structure and organization of an organ are based on a number of factors including growth, migration, elimination, and death of various cells; communication between cells to establish the correct architecture; competition between cells; and the composition of the extracellular matrix that is produced. In addition to normal cells stopping proliferation in an organ when that is appropriate, normal tissues have various mechanisms for eliminating cells both in the process of development as well as ongoing homeostasis of an organ. These include mechanical processes based on a number of factors including cell size, shape, and topology between cells that can determine cell fate as well as an active process of cell extrusion, which plays a major role in the elimination of both cells that are no longer needed by the organ and cells that are damaged and potentially dangerous (such as those with mutations that might be precursors for malignancy). The process of cell extrusion may depend on cell cycle arrest in the S phase; aberrations in this process may contribute to the metastatic process. A variety of processes, including major alterations in cell cycle control, apoptosis and other mechanisms of cell death, and uncontrolled cell signaling, all contribute to defects in appropriate cell extrusion contributing to the development of cancer.
_____
_____
Section-14
DNA damage and cancer:
DNA damage leads to mutagenesis and mutagenesis leads to Cancer.
This section deals with DNA damage and mutagenesis is discussed in next section.
_
DNA damage is considered to be the primary cause of cancer. More than 60,000 new naturally-occurring instances of DNA damage arise, on average, per human cell, per day, due to endogenous cellular processes. Additional DNA damage can arise from exposure to exogenous agents. Exogenous and endogenous agents that induce DNA damage have been identified as major causes of many common cancers. These include cancers of the lung (tobacco smoke), colorectum (exposure to bile acids that cause increased reactive oxygen species and reactive nitrogen species, and are produced in response to a high fat diet), esophagus (exposure to stomach acids plus bile acids due to gastroesophageal reflux), stomach (reactive oxygen species caused by Helicobacter pylori infection[), liver (Aspergillus metabolite aflatoxin B1), cervix/uterus (human papillomavirus plus increased nitric oxide from tobacco smoke or other infection) and melanoma (UV light from solar radiation). Inherited germ line mutations in DNA repair genes similarly cause an increase in DNA damages due to a deficiency in repair capability, and these also cause increases in cancer risk.
_
Generation of DNA damage (also known as DNA adducts or lesions) induced by various agents is an important first step in the process of carcinogenesis. Evolutionary processes gave rise to DNA repair tools that are efficient in repairing damaged DNA; yet replication of damaged DNA may take place prior to repair, particularly when they are induced at a high frequency. Damaged DNA replication may lead to gene mutations, which in turn may give rise to altered proteins. Mutations in an oncogene, a tumor-suppressor gene, or a gene that controls the cell cycle can generate a clonal cell population with a distinct advantage in proliferation. Many such events, broadly divided into the stages of initiation, promotion, and progression, which may occur over a long period of time and transpire in the context of chronic exposure to carcinogens, can lead to the induction of human cancer. This is exemplified in the long-term use of tobacco being responsible for an increased risk of lung cancer.
_
It is impossible to determine the initial cause for most specific cancers. In a few cases, only one cause exists: for example, the virus HHV-8 causes all Kaposi’s sarcomas. However, with the help of cancer epidemiology techniques and information, it is possible to produce an estimate of a likely cause in many more situations. For example, lung cancer has several causes, including tobacco use and radon gas. Men who currently smoke tobacco develop lung cancer at a rate 14 times that of men who have never smoked tobacco: the chance of lung cancer in a current smoker being caused by smoking is about 93%; there is a 7% chance that the smoker’s lung cancer was caused by radon gas or some other, non-tobacco cause. These statistical correlations have made it possible for researchers to infer that certain substances or behaviors are carcinogenic. Tobacco smoke causes increased exogenous DNA damage, and this DNA damage is the likely cause of lung cancer due to smoking. Among the more than 5,000 compounds in tobacco smoke, the genotoxic DNA-damaging agents that occur both at the highest concentrations, and which have the strongest mutagenic effects are acrolein, formaldehyde, acrylonitrile, 1,3-butadiene, acetaldehyde, ethylene oxide and isoprene.
_
Using molecular biological techniques, it is possible to characterize the mutations, epimutations or chromosomal aberrations within a tumor, and rapid progress is being made in the field of predicting certain cancer patients’ prognosis based on the spectrum of mutations. For example, up to half of all tumors have a defective p53 gene. This mutation is associated with poor prognosis, since those tumor cells are less likely to go into apoptosis or programmed cell death when damaged by therapy. Telomerase mutations remove additional barriers, extending the number of times a cell can divide. Other mutations enable the tumor to grow new blood vessels to provide more nutrients, or to metastasize, spreading to other parts of the body. However, once a cancer is formed it continues to evolve and to produce sub-clones. It was reported in 2012 that a single renal cancer specimen, sampled in nine different areas, had 40 “ubiquitous” mutations, found in all nine areas, 59 mutations shared by some, but not all nine areas, and 29 “private” mutations only present in one area.
_
The lineages of cells in which all these DNA alterations accumulate are difficult to trace, but two recent lines of evidence suggest that normal stem cells may be the cells of origin in cancers.
First, there exists a highly positive correlation (Spearman’s rho = 0.81; P < 3.5 × 10^−8) between the risk of developing cancer in a tissue and the number of normal stem cell divisions taking place in that same tissue. The correlation applied to 31 cancer types and extended across five orders of magnitude. This correlation means that if normal stem cells from a tissue divide once, the cancer risk in that tissue is approximately 1X. If they divide 1,000 times, the cancer risk is 1,000X. And if the normal stem cells from a tissue divide 100,000 times, the cancer risk in that tissue is approximately 100,000X. This strongly suggests that the main factor in cancer initiation is the fact that “normal” stem cells divide, which implies that cancer originates in normal, healthy stem cells.
Second, statistics show that most human cancers are diagnosed in older people. A possible explanation is that cancers occur because cells accumulate damage through time. DNA is the only cellular component that can accumulate damage over the entire course of a life, and stem cells are the only cells that can transmit DNA from the zygote to cells late in life. Other cells, derived from stem cells, do not keep DNA from the beginning of life until a possible cancer occurs. This implies that most cancers arise from normal stem cells.
_
DNA Damage and DNA Repair:
DNA damage occurs continuously in all organisms via a number of endogenous and exogenous factors, and it seems to play a central role in many biological processes, ultimately leading to cancer. Hence, robust DNA repair systems, which repair this damage, have evolved to maintain genomic integrity. The importance of DNA repair was underscored by conferring the Nobel Prize in Chemistry in 2015 to Tomas Lindahl, Paul Modrich, and Aziz Sancar for mapping, at a molecular level, how cells repair damaged DNA and protect the genetic information.
_
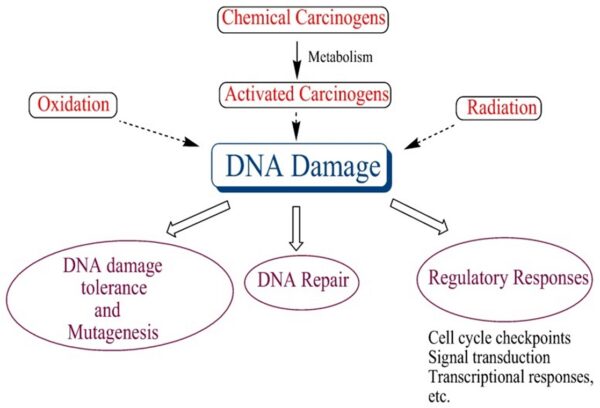
Figure above shows that DNA damage plays a central role in many biological processes linked to cancer.
DNA replication occurs during the S (synthetic) phase of cell cycle, which is preceded by the G1 (Gap 1) phase. The nuclear division occurs in the M (mitosis) phase, which takes place after the G2 phase. The differentiated cells at the G0 phase do not proliferate, whereas the G1, S, and G2 phases of a proliferating cell constitute the time lapse between two consecutive mitoses. The progression of a cell during cell cycle is regulated by cyclin dependent kinase in order to avoid the initiation of a cell cycle before the preceding one is completed. DNA damage interferes with the cell cycle, and therefore, there are checkpoint proteins that delay cell cycle progression providing the necessary time for DNA repair. If the DNA damage exceeds the capability of repair, pathways to trigger cell death are activated by apoptosis. The checkpoint pathways accordingly play an integral role in DNA damage response, and dysfunction of these pathways are important for the pathogenesis of malignant cells.
A deficiency in DNA repair would cause more DNA damage to accumulate, and increase the risk for cancer. For example, individuals with an inherited impairment in any of 34 DNA repair genes are at increased risk of cancer, with some defects causing an up to 100% lifetime chance of cancer. These 34 inherited human DNA repair gene mutations increase cancer risk, including, for example, germ line mutations in the BRCA1, XPC and MLH1 genes.
_
Relationship between DNA Adducts and Tumor Incidence:
Carcinogens and mutagens usually generate multiple DNA adducts, and it was shown that certain adducts are biologically more relevant than others. Many diseases in humans are the result of specific genetic mutations. Therefore, DNA adducts or lesions that lead to mutations became the focus of many studies. As for example, the predominant mutation induced by most methylating and ethylating agents are G:C→A:T transitions induced by O6-alkylguanine, even though the major adduct is formed at the N7-position of guanine.
Characterization of a quantitative relationship between DNA adduct levels and tumor incidences in rats and mice was attempted by Ottender and Lutz. Of the 27 different chemicals investigated, the range of carcinogenic potency of structurally different DNA adducts is typically within 2 orders of magnitude. In the rat, for instance, 53 adducts per 108 nucleotides for the aflatoxin B1 to 2082 adducts per 108 nucleotides for dimethylnitrosamine relate to the normalized 50% level of liver tumor incidences, suggesting that the aflatoxin–DNA adducts are 40 times more potent than the adducts formed by dimethylnitrosamine for inducing hepatocellular carcinoma.
_
Damaged DNA Replication:
DNA replication causes mutations and DNA damage, or DNA adducts increases the rate of error-prone replication. However, each DNA damage or adduct has a unique mutational signature, which is directly related to the identity of the DNA polymerase that bypass it and the mechanism of its nucleotide insertion and extension.
A human cell contains at least 17 different DNA polymerases. The DNA polymerases belong to seven families (A, B, C, D, X, Y, and RT), of which the C family enzymes were only found in prokaryotes. In eukaryotes, the B-family enzymes pol ε and pol δ carry out a large fraction of nuclear DNA replication, whereas pol α of the same family performs initiation and priming. These three polymerases are essential for DNA replication in eukaryotes. In the current model of DNA replication, pol ε carries out majority of leading strand DNA replication of the undamaged genome, whereas pol δ primarily replicates the lagging strand. But this model has recently been challenged, and data supporting involvement of pol δ in both leading and lagging strand replication have been presented. It is noteworthy that these important DNA polymerases are inefficient in bypassing most bulky or distorting DNA damages, such as the ones induced by PAHs and UV light.
The discovery of translesion synthesis (TLS) DNA polymerases in the 1990s and the study of their catalytic and non-catalytic roles in damaged DNA replication provided much of our current understanding of DNA adduct or lesion bypass. Lesion bypass is carried out primarily by the Y-family polymerases. But X- and B-family polymerases are also involved in many cases.
TLS of various types of DNA damage have been conducted by genetic studies in repair and replication competent cells, by in vitro experiments using purified DNA polymerases and accessory proteins, and by structural and computational studies. The mechanistic information gathered from these studies is critical to understand the mechanism of mutagenesis, the underlying process for the development of cancer. These fundamental studies are now allowing therapeutic application, as inhibiting the activity of some of the TLS polymerases may enhance the effect of an antitumor agent.
_____
_____
DNA damages cause epigenetic changes and mutations:
Figure below depicts central role of DNA damage and epigenetic defects in DNA repair genes in carcinogenesis.
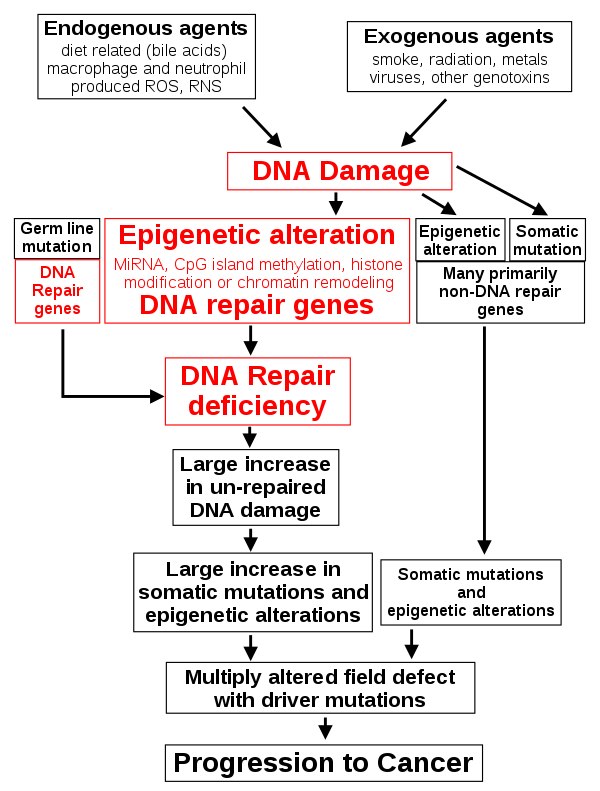
Exogenous and endogenous sources of DNA damage are indicated in the boxes at the top of the figure in this section. The central role of DNA damage in progression to cancer is indicated at the second level of the figure. The central elements of DNA damage, epigenetic alterations and deficient DNA repair in progression to cancer are shown in red. Germline mutations are shown in a box at the left of the figure, with an indication of their contribution to DNA repair deficiency. However, such germline mutations (which cause highly penetrant cancer syndromes) are the cause of only about one percent of cancers. In sporadic cancers, a deficiency in DNA repair is occasionally due to a mutation in a DNA repair gene; much more frequently, reduced or absent expression of DNA repair genes is due to epigenetic alterations that reduce or silence gene expression. This is indicated in the figure at the 3rd level from the top. For example, for 113 colorectal cancers examined in sequence, only four had a missense mutation in the DNA repair gene MGMT, while the majority had reduced MGMT expression due to methylation of the MGMT promoter region (an epigenetic alteration).
_
ROS, produced during inflammation and other types of cellular stress, cause a variety of types of DNA damage, some of which lead to double strand breaks. During repair of double strand breaks and other types of oxidative DNA damages, methylation of promoter CpG islands in DNA and/or modification of histones can occur, causing gene silencing. These epigenetic alterations are sometimes not reversed after repair is completed. While it has long been known that oxidative damage can cause mutation, it has only recently become clear that oxidative damage can also give rise to epigenetic changes (epimutation).
Other types of DNA damage can also give rise to epimutation during DNA repair. The DNA repair enzyme Parp1 [poly(ADP)-ribose polymerase-1] acts at sites of DNA damage, especially single strand breaks, where it adds poly(ADP)-ribose to specific proteins as part of the overall DNA repair process. This, in turn, directs recruitment and activation of the chromatin remodeling protein ALC1 to cause nucleosome remodeling. Nucleosome remodeling has been found to cause, for instance, epigenetic silencing of DNA repair gene MLH1. In addition, certain chemicals previously identified as DNA damaging agents, including benzene, hydroquinone, styrene, carbon tetrachloride and trichloroethylene, cause considerable hypomethylation of DNA, leading to epigenetic modifications, and some of this hypomethylation occurs through the activation of oxidative stress pathways.
_
Epigenetic changes in DNA repair gene expression are a likely source of genomic instability:
While germ line (familial) mutations in DNA repair genes cause a high risk of cancer, in sporadic (non-familial) cancers, by contrast, somatic mutations in DNA repair genes are rarely found. However, deficient expression of DNA repair genes is frequently observed within sporadic cancers, and this is almost always due to epigenetic alteration. Epimutation leading to silencing of a gene necessary for DNA repair will allow unrepaired damages to increase. Such additional DNA damages, in turn, will cause increased mutations and epimutations, including carcinogenic driver mutations and epimutations.
Truninger et al compared the frequencies of germ line mutations, CpG island methylations and other unidentified alterations in the down-regulation of expression of DNA mismatch repair (MMR) gene MLH1 in colon cancer. They evaluated 1048 unselected consecutive colon cancers. They found that 103 of these cancers were deficient in protein expression of MLH1. Among the MLH1 deficient cancers, 68 were sporadic and the remaining 35 were due to germ line mutations. Among the 68 sporadic MLH1 protein-deficient colon cancers, 65 (96%) were deficient due to epigenetic methylation of the CpG island of the MLH1 gene. Reduced protein expression of MLH1 in the remaining 3 sporadic MLH1 protein-deficient cancers may have been caused by over expression of the microRNA miR-155. This explanation is suggested by the finding that transfection of miR-155 into cells caused reduced expression of MLH1. Furthermore, high expression of miR-155 was found in colon cancers in which protein expression of MLH1 was reduced and the MLH1 gene was neither mutated nor hypermethylated.
__
Contribution of field defects:
The somatic mutations and epigenetic alterations caused by DNA damage and deficiencies in DNA repair accumulate in field defects. Field defects are normal-appearing tissues with multiple alterations, and are common precursors to development of the disordered and over-proliferating clone of tissue in a cancer. Such field defects may have numerous mutations and epigenetic alterations.
Figure below shows longitudinally opened freshly resected colon segment showing a cancer and four polyps. Plus a schematic diagram indicating a likely field defect (a region of tissue that precedes and predisposes to the development of cancer) in this colon segment. The diagram indicates sub-clones and sub-sub-clones that were precursors to the tumors.
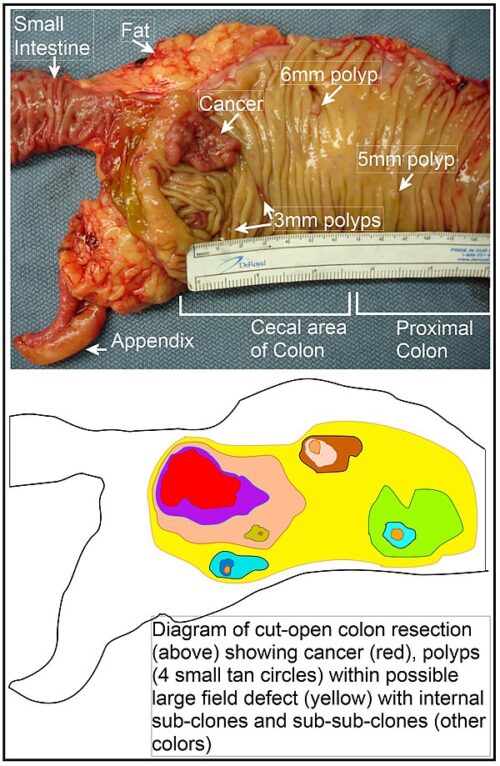
_
The term “field cancerization” was first used in 1953 to describe an area or “field” of epithelium that has been preconditioned by (at that time) largely unknown processes so as to predispose it towards development of cancer. Since then, the terms “field cancerization” and “field defect” have been used to describe pre-malignant tissue in which new cancers are likely to arise. Field defects have been identified in association with cancers and are important in progression to cancer. However, it was pointed out by Rubin that “the vast majority of studies in cancer research has been done on well-defined tumors in vivo, or on discrete neoplastic foci in vitro. Yet there is evidence that more than 80% of the somatic mutations found in mutator phenotype human colorectal tumors occur before the onset of terminal clonal expansion…” More than half of somatic mutations identified in tumors occurred in a pre-neoplastic phase (in a field defect), during growth of apparently normal cells. It would also be expected that many of the epigenetic alterations present in tumors may have occurred in pre-neoplastic field defects.
_
In the colon, a field defect probably arises by natural selection of a mutant or epigenetically altered cell among the stem cells at the base of one of the intestinal crypts on the inside surface of the colon. A mutant or epigenetically altered stem cell may replace the other nearby stem cells by natural selection. This may cause a patch of abnormal tissue to arise. The figure above includes a photo of a freshly resected and lengthwise-opened segment of the colon showing a colon cancer and four polyps. Below the photo there is a schematic diagram of how a large patch of mutant or epigenetically altered cells may have formed, shown by the large area in yellow in the diagram. Within this first large patch in the diagram (a large clone of cells), a second such mutation or epigenetic alteration may occur, so that a given stem cell acquires an advantage compared to its neighbors, and this altered stem cell may expand clonally, forming a secondary patch, or sub-clone, within the original patch. This is indicated in the diagram by four smaller patches of different colors within the large yellow original area. Within these new patches (sub-clones), the process may be repeated multiple times, indicated by the still smaller patches within the four secondary patches (with still different colors in the diagram) which clonally expand, until stem cells arise that generate either small polyps or else a malignant neoplasm (cancer). In the photo, an apparent field defect in this segment of a colon has generated four polyps (labeled with the size of the polyps, 6mm, 5mm, and two of 3mm, and a cancer about 3 cm across in its longest dimension). These neoplasms are also indicated (in the diagram below the photo) by 4 small tan circles (polyps) and a larger red area (cancer). The cancer in the photo occurred in the caecal area of the colon, where the colon joins the small intestine (labeled) and where the appendix occurs (labeled). The fat in the photo is external to the outer wall of the colon. In the segment of colon shown here, the colon was cut open lengthwise to expose its inner surface and to display the cancer and polyps occurring within the inner epithelial lining of the colon.
If the general process by which sporadic colon cancers arise is the formation of a pre-neoplastic clone that spreads by natural selection, followed by formation of internal sub-clones within the initial clone, and sub-sub-clones inside those, then colon cancers generally should be associated with, and be preceded by, fields of increasing abnormality, reflecting the succession of premalignant events. The most extensive region of abnormality (the outermost yellow irregular area in the diagram) would reflect the earliest event in formation of a malignant neoplasm.
_
In experimental evaluation of specific DNA repair deficiencies in cancers, many specific DNA repair deficiencies were also shown to occur in the field defects surrounding those cancers. The table below gives examples for which the DNA repair deficiency in a cancer was shown to be caused by an epigenetic alteration, and the somewhat lower frequencies with which the same epigenetically caused DNA repair deficiency was found in the surrounding field defect.
Frequency of epigenetic changes in DNA repair genes in sporadic cancers and in adjacent field defects:
|
Cancer |
Gene |
Frequency in Cancer |
Frequency in Field Defect |
|
Colorectal |
MGMT |
46% |
34% |
|
Colorectal |
MGMT |
47% |
11% |
|
Colorectal |
MGMT |
70% |
60% |
|
Colorectal |
MSH2 |
13% |
5% |
|
Colorectal |
ERCC1 |
100% |
40% |
|
Colorectal |
PMS2 |
88% |
50% |
|
Colorectal |
XPF |
55% |
40% |
|
Head and Neck |
MGMT |
54% |
38% |
|
Head and Neck |
MLH1 |
33% |
25% |
|
Head and Neck |
MLH1 |
31% |
20% |
|
Stomach |
MGMT |
88% |
78% |
|
Stomach |
MLH1 |
73% |
20% |
|
Esophagus |
MLH1 |
77%–100% |
23%–79% |
_____
_____
Section-15
Mutations and cancer:
_
Heredity (Germline) Versus Acquired (Somatic) Mutations:
Talking about mutations and cancer can be confusing because there are two different types of mutations to consider.
Germline mutations:
Hereditary or germline mutations are gene mutations that are present at birth and exist in all of the cells of the body. Examples of germline mutations are those in the BRCA genes (tumor suppressor genes) and non-BRCA genes that increase the risk of developing breast cancer.
Mutation of tumor suppressor genes that are passed on to the next generation of not merely cells, but their offspring, can cause increased likelihoods for cancers to be inherited. Members within these families have increased incidence and decreased latency of multiple tumors. The mode of inheritance of mutant tumor suppressors is that affected member inherits a defective copy from one parent, and a normal copy from another. Because mutations in tumor suppressors act in a recessive manner (note, however, there are exceptions), the loss of the normal copy creates the cancer phenotype. For instance, individuals that are heterozygous for p53 mutations are often victims of Li-Fraumeni syndrome, and that are heterozygous for Rb mutations develop retinoblastoma. In similar fashion, mutations in the adenomatous polyposis coli gene are linked to adenopolyposis colon cancer, with thousands of polyps in the colon while young, whereas mutations in BRCA1 and BRCA2 lead to early onset of breast cancer.
Somatic Mutations:
Somatic or acquired mutations, in contrast, are those that occur after birth and are not passed down from one generation to another (not hereditary). These mutations are not present in all cells, but rather occur in a particular type of cell in the process of that cell becoming malignant or cancerous. Many of the targeted therapies used to treat cancer are designed to address changes in cell growth caused by these particular mutations.
_
Current evidence indicates that the great majority of human cancers are caused by somatic mutations in certain specific genes. Genes with this property are called “oncogenes”, and more than 70 kinds of oncogenes are now known. Based on the increasing frequency with which new methods for finding oncogenes have been finding the same ones over and over again, it is estimated there are upwards of 100 different kinds of oncogenes, total!
Somatic mutations, of course, are any changes in the genetic information in somatic cells (i.e. cells other than sperm or egg cell precursors). Somatic mutations are very common (billions of them occur in a normal person); they are passed down only to the daughter cells of the cell undergoing the original mutation, and to their daughter’s daughter cells, etc. thus forming a clone of genetically different cells within the body. Nearly all somatic mutations are harmless, either with no effect or making the affected cells slightly abnormal (defective but not dangerous).
But it is very, very dangerous to over-express or otherwise change that tiny minority of genes that code for the proteins whose function is to control cell growth. Cancer can result from single base changes in some of these genes, but is more frequently caused by gene duplications, deletions, or by translocations of genes from their normal location. Translocation to a position adjacent to the control regions (promoters and/or enhancers) of other genes can cause the translocated genes to be ‘turned on’ (transcribed) when they shouldn’t be, or more than they should be. This causes most lymphomas.
_
Almost all cancers are believed to be clonal in the sense that all the cells of a given person’s cancer are descended from just one original somatic cell that underwent a mutation in one (or more) oncogenes. The evidence includes such facts as that, in female mammals (where only one of the two X chromosomes is active in any given cell, with half the somatic cells having one of the Xs active and the other having the other X active) all the cells of any given tumor have the same active X. Likewise, cancers in chimeric animals (such as tetraparental mice) are found to contain only cells derived from one of the original embryos from which the animal was formed. In other words, cancer does not spread from cell to cell; the enlargement of tumors does not involve any kind of recruitment of previously normal cells; and it is not “catching”.
_
Carcinogenesis, also called oncogenesis or tumorigenesis, is the formation of a cancer, whereby normal cells are transformed into cancer cells. The process is characterized by changes at the cellular, genetic, and epigenetic levels and abnormal cell division. Cell division is a physiological process that occurs in almost all tissues and under a variety of circumstances. Normally, the balance between proliferation and programmed cell death, in the form of apoptosis, is maintained to ensure the integrity of tissues and organs. According to the prevailing accepted theory of carcinogenesis, the somatic mutation theory, mutations in DNA and epimutations that lead to cancer disrupt these orderly processes by interfering with the programming regulating the processes, upsetting the normal balance between proliferation and cell death. This results in uncontrolled cell division and the evolution of those cells by natural selection in the body. Only certain mutations lead to cancer whereas the majority of mutations do not.
_
Variants of inherited genes may predispose individuals to cancer. In addition, environmental factors such as carcinogens and radiation cause mutations that may contribute to the development of cancer. Finally random mistakes in normal DNA replication may result in cancer causing mutations. A series of several mutations to certain classes of genes is usually required before a normal cell will transform into a cancer cell. Recent comprehensive patient-level classification and quantification of driver events in TCGA cohorts revealed that there are on average 12 driver events per tumor, of which 0.6 are point mutations in oncogenes, 1.5 are amplifications of oncogenes, 1.2 are point mutations in tumor suppressors, 2.1 are deletions of tumor suppressors, 1.5 are driver chromosome losses, 1 is a driver chromosome gain, 2 are driver chromosome arm losses, and 1.5 are driver chromosome arm gains. Mutations in genes that regulate cell division, apoptosis (cell death), and DNA repair may result in uncontrolled cell proliferation and cancer.
_
Cancer is fundamentally a disease of regulation of tissue growth. In order for a normal cell to transform into a cancer cell, genes that regulate cell growth and differentiation must be altered. Genetic and epigenetic changes can occur at many levels, from gain or loss of entire chromosomes, to a mutation affecting a single DNA nucleotide, or to silencing or activating a microRNA that controls expression of 100 to 500 genes. There are two broad categories of genes that are affected by these changes. Oncogenes may be normal genes that are expressed at inappropriately high levels, or altered genes that have novel properties. In either case, expression of these genes promotes the malignant phenotype of cancer cells. Tumor suppressor genes are genes that inhibit cell division, survival, or other properties of cancer cells. Tumor suppressor genes are often disabled by cancer-promoting genetic changes. Finally, Oncovirinae, viruses that contain an oncogene, are categorized as oncogenic because they trigger the growth of tumorous tissues in the host. This process is also referred to as viral transformation.
_
Cancers and tumors are caused by a series of mutations as seen in the figure below. Each mutation alters the behavior of the cell somewhat.
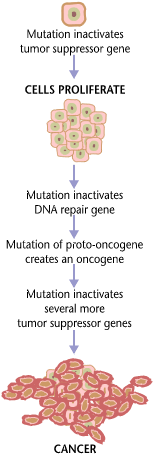
Multiple mutations exist in cancer cells. For example, a mutation limited to one oncogene would be suppressed by normal mitosis control and tumor suppressor genes. A mutation to only one tumor suppressor gene would not cause cancer either, due to the presence of many “backup” genes that duplicate its functions. It is only when enough proto-oncogenes have mutated into oncogenes, and enough tumor suppressor genes deactivated or damaged, that the signals for cell growth overwhelm the signals to regulate it, that cell growth quickly spirals out of control. Often, because these genes regulate the processes that prevent most damage to genes themselves, the rate of mutations increases as one gets older, because DNA damage forms a feedback loop.
_____
_____
Several types of cancers are the result of at least a few mutations in critical genes. The somatic mutation theory (SMT) of cancer, the most prevalent model, proposes that cancer is caused by mutation(s) in the body cells (as opposed to germ cells), especially nonlethal mutations associated with increased proliferation of the mutant cells. The SMT hypothesis originated from Theodore Boveri’s postulate in 1914 that a combination of chromosomal defects could result in cancer. After Watson and Crick’s discovery of the structure of DNA that also implied that DNA contains the genetic information, in 1953, Carl O. Nordling proposed that several mutated genes may lead to cancer. Ashley suggested that cancer may occur as a result of three to seven mutations. Alfred Knudson modified Ashley’s proposal, based on his observations of a number of retinoblastoma cases, proposing that cancer is the result of accumulated mutations to a cell’s DNA, which could be as little as two hits. The two-hit model proposes that dominantly inherited predisposition to cancer requires a germline mutation, while tumorigenesis necessitates a second somatic mutation. For colorectal carcinoma, Fearon and Vogelstein suggested that four to five gene mutations are necessary for the development of malignant tumor, and the accumulation of the mutations, rather than their specific order, is the critical determinant of tumorigenesis. More recently, these mutations have been referred to as “driver” mutations conferring growth advantage to the cells. In humans, more than 350 mutated genes that are implicated in the development of cancer have been identified. A large-scale sequencing study has shown that most somatic mutations in cancer cells are “passengers” that do not cause tumorigenesis, whereas 120 of the 518 genes screened (~23%) carry a “driver” mutation, which can function as cancer genes. Similar conclusions have been reached in other studies. The basic premise of SMT, however, has been challenged from time to time. A new idea announced in 2011 is an extreme version of multiple mutations, called chromothripsis by its proponents. This idea, affecting only 2–3% of cases of cancer, although up to 25% of bone cancers, involves the catastrophic shattering of a chromosome into tens or hundreds of pieces and then being patched back together incorrectly. This shattering probably takes place when the chromosomes are compacted during normal cell division, but the trigger for the shattering is unknown. Under this model, cancer arises as the result of a single, isolated event, rather than the slow accumulation of multiple mutations.
_
Genes that contribute to cancer include oncogenes and tumor suppressor genes. Oncogenes change a normal healthy cell into a cancerous cell. Examples include the ras family of genes and HER2. The ras genes produce proteins engaged in cell communication pathways, cell growth, and cell death, whereas HER2 makes specialized proteins controlling cell growth, and spread notably in breast and ovarian cancer cells. DNA adduct-induced mutations in the ras gene, at the activating codons 12, 13, 59, and 61, are considered to be of note. Aflatoxin B1 (AFB1) causes G·C→A·T or G·C→T·A substitutions at codon 12 in experimental animals. Analyses of lung tumors in A/J mice by the tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) and related compounds showed high frequency of G→A mutations (GGT to GAT) in codon 12. By contrast, tumor suppressor genes protect a cell from becoming cancerous. The tumor suppressor proteins control cell growth by monitoring cell division, repairing base mismatches in DNA, and controlling cell death (apoptosis). Examples of tumor suppressor genes include p53, BRCA1, and BRCA2. More than 50% of human cancers are characterized by mutations in the p53 gene, and most p53 gene mutations are not hereditary. Germline mutations in BRCA1 or BRCA2 gene increases a woman’s risk of hereditary breast and ovarian cancer. A convincing relationship between a chemical and p53 mutation in human cancer has been shown in geographical areas where AFB1-derived liver cancers accompanied unusually high frequency of G·C→T·A mutations at the third base of codon 249 of the p53 gene. Also, a human liver cell line following exposure to AFB1 showed the same mutation at the third base of p53 codon 249. Likewise, for lung cancer cases of smokers, ~40% of the mutations involved G→T transversions, and more than 90% of them are on a guanine on the non-transcribed strand. Major hotspots are observed at codons 157, 248, and 273. Even though codon 157 is unique to lung cancer, the other two are hotspots for mutations in many other cancers, usually detected as transitions at these CpG sequences, whereas in lung cancers, G→T transversions are the most common mutations. Pfeifer and colleagues have claimed that sequence specificity of G→T transversions in lung tumors is consistent with a direct mutagenic action of Polycyclic aromatic hydrocarbons (PAHs) compounds, such as B[a]P present in cigarette smoke. In addition to the cancers induced by exogenous agents, few hereditary cancers, which include retinoblastoma and Li-Fraumeni syndrome, involve germline mutations in tumor suppressor genes.
_
Human tumors are largely heterogeneous. Loeb and coworkers suggest that this heterogeneity results from a mutator phenotype. They hypothesized that increased mutation rates are essential to account for the large number of mutations observed in cancer cells. Consequently, an initial mutator mutation triggers additional mutations, including mutations in genes that maintain genetic stability, starting a cascade of mutations throughout the genome. Several types of cancers exhibit mutator phenotype resulting from mutations at loci responsible for DNA mismatch repair. It was also proposed that p53 mutations might give rise to mutator phenotype, because p53 is a gatekeeper of DNA damage responses. However, others believe that a mutator phenotype is not necessary for tumor initiation and progression, in spite of the fact that some tumors may acquire it during tumorigenesis.
____
____
Non-mutagenic carcinogens:
Carcinogens are currently categorized into two classes, genotoxic and non-genotoxic carcinogens, which are subject to different regulatory policies. Genotoxic carcinogens are chemicals that exert carcinogenicity via the induction of mutations. Owing to their DNA interaction properties, there is thought to be no safe exposure threshold or dose. Genotoxic carcinogens are regulated under the assumption that they pose a cancer risk for humans, even at very low doses. In contrast, non-genotoxic carcinogens, which induce cancer through mechanisms other than mutations, such as hormonal effects, cytotoxicity, cell proliferation, or epigenetic changes, are thought to have a safe exposure threshold or dose; thus, their use in society is permitted unless the exposure or intake level would exceed the threshold. In the majority of instances, chemical carcinogens, directly or after metabolism, induce DNA damage and act in a ‘genotoxic’ manner. There is, however, a group of carcinogens that induce cancer via non-genotoxic mechanisms. Non-genotoxic carcinogens have been shown to act as tumor promoters (1,4-dichlorobenzene), endocrine-modifiers (17beta-estradiol), receptor-mediators (2,3,7,8-tetrachlorodibenzo-p-dioxin), immunosuppressants (cyclosporine) or inducers of tissue-specific toxicity and inflammatory responses (metals such as arsenic and beryllium). The diversity of modes of action of non-genotoxic carcinogens, the tissue and species specificity, and the absence of genotoxicity makes predicting their carcinogenic potential extremely challenging.
______
______
Cancer Genome Landscapes, a 2013 study:
Over the past decade, comprehensive sequencing efforts have revealed the genomic landscapes of common forms of human cancer. For most cancer types, this landscape consists of a small number of “mountains” (genes altered in a high percentage of tumors) and a much larger number of “hills” (genes altered infrequently). To date, these studies have revealed ~140 genes that, when altered by intragenic mutations, can promote or “drive” tumorigenesis. A typical tumor contains two to eight of these “driver gene” mutations; the remaining mutations are passengers that confer no selective growth advantage. Driver genes can be classified into 12 signaling pathways that regulate three core cellular processes: cell fate, cell survival, and genome maintenance. A better understanding of these pathways is one of the most pressing needs in basic cancer research.
_
How many genes are subtly mutated in a typical Human Cancer?
In common solid tumors such as those derived from the colon, breast, brain, or pancreas, an average of 33 to 66 genes display subtle somatic mutations that would be expected to alter their protein products. About 95% of these mutations are single-base substitutions (such as C>G), whereas the remainder are deletions or insertions of one or a few bases (such as CTT>CT). Of the base substitutions, 90.7% result in missense changes, 7.6% result in nonsense changes, and 1.7% result in alterations of splice sites or untranslated regions immediately adjacent to the start and stop codons.
_
Mutation Timing:
When do these mutations occur?
Tumors evolve from benign to malignant lesions by acquiring a series of mutations over time, a process that has been particularly well studied in colorectal tumors. The first, or “gatekeeping,” mutation provides a selective growth advantage to a normal epithelial cell, allowing it to outgrow the cells that surround it and become a microscopic clone. Gatekeeping mutations in the colon most often occur in the APC gene. The small adenoma that results from this mutation grows slowly, but a second mutation in another gene, such as KRAS, unleashes a second round of clonal growth that allows an expansion of cell number. The cells with only the APC mutation may persist, but their cell numbers are small compared with the cells that have mutations in both genes. This process of mutation followed by clonal expansion continues, with mutations in genes such as PIK3CA, SMAD4, and TP53, eventually generating a malignant tumor that can invade through the underlying basement membrane and metastasize to lymph nodes and distant organs such as the liver. The mutations that confer a selective growth advantage to the tumor cell are called “driver” mutations. It has been estimated that each driver mutation provides only a small selective growth advantage to the cell, on the order of a 0.4% increase in the difference between cell birth and cell death. Over many years, however, this slight increase, compounded once or twice per week, can result in a large mass, containing billions of cells.
_
The number of mutations in certain tumors of self-renewing tissues is directly correlated with age. When evaluated through linear regression, this correlation implies that more than half of the somatic mutations identified in these tumors occur during the preneoplastic phase; that is, during the growth of normal cells that continuously replenish gastrointestinal and genito-urinary epithelium and other tissues. All of these pre-neoplastic mutations are “passenger” mutations that have no effect on the neoplastic process. This result explains why a colorectal tumor in a 90-year-old patient has nearly twice as many mutations as a morphologically identical colorectal tumor in a 45-year-old patient. This finding also partly explains why advanced brain tumors (glioblastomas) and pancreatic cancers (pancreatic ductal adenocarcinomas) have fewer mutations than colorectal tumors; glial cells of the brain and epithelial cells of the pancreatic ducts do not replicate, unlike the epithelial cells lining the crypts of the colon. Therefore, the gatekeeping mutation in a pancreatic or brain cancer is predicted to occur in a precursor cell that contains many fewer mutations than are present in a colorectal precursor cell. This line of reasoning also helps to explain why pediatric cancers have fewer mutations than adult tumors. Pediatric cancers often occur in non–self-renewing tissues, and those that arise in renewing tissues (such as leukemia) originate from precursor cells that have not renewed themselves as often as in adults. In addition, pediatric tumors, as well as adult leukemia and lymphomas, may require fewer rounds of clonal expansion than adult solid tumors. Genome sequencing studies of leukemia patients support the idea that mutations occur as random events in normal precursor cells before these cells acquire an initiating mutation.
_
When during tumorigenesis do the remaining somatic mutations occur?
Because mutations in tumors occur at predictable and calculable rates, the number of somatic mutations in tumors provides a clock, much like the clock used in evolutionary biology to determine species divergence time. The number of mutations has been measured in tumors representing progressive stages of colorectal and pancreatic cancers. Applying the evolutionary clock model to these data leads to two unambiguous conclusions: First, it takes decades to develop a full-blown, metastatic cancer. Second, virtually all of the mutations in metastatic lesions were already present in a large number of cells in the primary tumors.
_
The timing of mutations is relevant to our understanding of metastasis, which is responsible for the death of most patients with cancer. The primary tumor can be surgically removed, but the residual metastatic lesions—often undetectable and widespread—remain and eventually enlarge, compromising the function of the lungs, liver, or other organs. From a genetics perspective, it would seem that there must be mutations that convert a primary cancer to a metastatic one, just as there are mutations that convert a normal cell to a benign tumor, or a benign tumor to a malignant one. Despite intensive effort, however, consistent genetic alterations that distinguish cancers that metastasize from cancers that have not yet metastasized remain to be identified.
One potential explanation invokes mutations or epigenetic changes that are difficult to identify with current technologies. Another explanation is that metastatic lesions have not yet been studied in sufficient detail to identify these genetic alterations, particularly if the mutations are heterogeneous in nature. But another possible explanation is that there are no metastasis genes. A malignant primary tumor can take many years to metastasize, but this process is, in principle, explicable by stochastic processes alone. Advanced tumors release millions of cells into the circulation each day, but these cells have short half-lives, and only a miniscule fraction establish metastatic lesions. Conceivably, these circulating cells may, in a nondeterministic manner, infrequently and randomly lodge in a capillary bed in an organ that provides a favorable microenvironment for growth. The bigger the primary tumor mass, the more likely that this process will occur. In this scenario, the continual evolution of the primary tumor would reflect local selective advantages rather than future selective advantages. The idea that growth at metastatic sites is not dependent on additional genetic alterations is also supported by recent results showing that even normal cells, when placed in suitable environments such as lymph nodes, can grow into organoids, complete with a functioning vasculature.
_
Other Types of Genetic Alterations in Tumors:
Though the rate of point mutations in tumors is similar to that of normal cells, the rate of chromosomal changes in cancer is elevated. Therefore, most solid tumors display widespread changes in chromosome number (aneuploidy), as well as deletions, inversions, translocations, and other genetic abnormalities. When a large part of a chromosome is duplicated or deleted, it is difficult to identify the specific “target” gene(s) on the chromosome whose gain or loss confers a growth advantage to the tumor cell. Target genes are more easily identified in the case of chromosome translocations, homozygous deletions, and gene amplifications. Translocations generally fuse two genes to create an oncogene (such as BCR-ABL in chronic myelogenous leukemia) but, in a small number of cases, can inactivate a tumor suppressor gene by truncating it or separating it from its promoter. Homozygous deletions often involve just one or a few genes, and the target is always a tumor suppressor gene. Amplifications contain an oncogene whose protein product is abnormally active simply because the tumor cell contains 10 to 100 copies of the gene per cell, compared with the two copies present in normal cells.
_
Most solid tumors have dozens of translocations; however, as with point mutations, the majority of translocations appear to be passengers rather than drivers. The breakpoints of the translocations are often in “gene deserts” devoid of known genes, and many of the translocations and homozygous deletions are adjacent to fragile sites that are prone to breakage. Cancer cells can, perhaps, survive such chromosome breaks more easily than normal cells because they contain mutations that incapacitate genes like TP53, which would normally respond to DNA damage by triggering cell death. Studies to date indicate that there are roughly 10 times fewer genes affected by chromosomal changes than by point mutations.
_
Drivers Versus Passenger Mutations:
Though it is easy to define a “driver gene mutation” in physiologic terms (as one conferring a selective growth advantage), it is more difficult to identify which somatic mutations are drivers and which are passengers. Moreover, it is important to point out that there is a fundamental difference between a driver gene and a driver gene mutation. A driver gene is one that contains driver gene mutations. But driver genes may also contain passenger gene mutations. For example, APC is a large driver gene, but only those mutations that truncate the encoded protein within its N-terminal 1600 amino acids are driver gene mutations. Missense mutations throughout the gene, as well as protein-truncating mutations in the C-terminal 1200 amino acids, are passenger gene mutations.
Further complicating matters, there are two distinct meanings of the term “driver gene” that are used in the cancer literature. The driver-versus-passenger concept was originally used to distinguish mutations that caused a selective growth advantage from those that did not. According to this definition, a gene that does not harbor driver gene mutations cannot be a driver gene. But many genes that contain few or no driver gene mutations have been labeled driver genes in the literature. These include genes that are overexpressed, underexpressed, or epigenetically altered in tumors, or those that enhance or inhibit some aspect of tumorigenicity when their expression is experimentally manipulated. Though a subset of these genes may indeed play an important role in the neoplastic process, it is confusing to lump them all together as driver genes.
To reconcile the two connotations of driver genes, authors suggest that genes suspected of increasing the selective growth advantage of tumor cells be categorized as either “Mut-driver genes” or “Epi-driver genes.” Mut-driver genes contain a sufficient number or type of driver gene mutations to unambiguously distinguish them from other genes. Epi-driver genes are expressed aberrantly in tumors but not frequently mutated; they are altered through changes in DNA methylation or chromatin modification that persist as the tumor cell divides.
______
______
Section-16
Genetics and epigenetics of cancer:
_
Gene activity can be changed by mutation or other ways (epimutations).
Gene mutations can be either inherited or acquired.
An inherited gene mutation, as the name implies, is inherited from a parent, so it’s present in the very first cell (once the egg cell is fertilized by a sperm cell) that eventually becomes a person. Since all the cells in the body came from this first cell, this mutation is in every cell in the body, and can also be passed on to the next generation. This type of mutation is also called a germline mutation (because the cells that develop into eggs and sperm are called germ cells) or a hereditary mutation.
It typically takes more than one gene mutation for a cell to become a cancer cell. But when someone inherits an abnormal copy of a gene, their cells already start out with one mutation. This makes it easier (and quicker) for other mutations to happen, which can lead to a cell becoming a cancer cell. This is why cancers related to inherited mutations tend to occur earlier in life than cancers of the same type that are not inherited. Inherited gene mutations are not the main cause of most cancers.
An acquired gene mutation is not inherited from a parent. Instead, it develops at some point during a person’s life. Acquired mutations occur in one cell, and then are passed on to any new cells that come from that cell. This mutation cannot be passed on to a person’s children, because it doesn’t affect their sperm or egg cells. This type of mutation is also called a sporadic mutation or a somatic mutation. Acquired mutations can happen for different reasons. Sometimes they happen when a cell’s DNA is damaged, such as after being exposed to radiation or certain chemicals. But often these mutations occur randomly, without having an outside cause. For example, during the complex process when a cell divides to make 2 new cells, the cell must make another copy of all of its DNA, and sometimes mistakes (mutations) occur while this is happening. Every time a cell divides is another chance for gene mutations to occur. The number of mutations in our cells can build up over time, which is why we have a higher risk of cancer as we get older. Acquired gene mutations are a much more common cause of cancer than inherited mutations.
_
Other ways gene activity can be changed:
Some of the changes inside cells that can lead to cancer don’t involve gene variants or mutations. Cells can turn some of their genes on and off in other ways, and some of these might also affect how a cell grows and divides. Different genes are more active in some cells than in others. Even within a certain cell, some genes are active at some times and inactive at others. Turning these genes on and off isn’t done by changing the DNA sequence (as is the case with variants and mutations). Instead, the changes in gene activity occur by other means known as epigenetic changes. There are several types of these changes:
- DNA methylation: In this type of change, a small chemical group called a methyl group is attached to the DNA so that the gene can’t start the process of making the protein it codes for. This basically turns off the gene. On the other hand, removing the methyl group (in a process called demethylation) can turn a gene on.
- Histone acetylation/histone modification: Chromosomes are made up of strands of DNA wrapped around proteins called histones. Histone proteins can be changed by adding (or subtracting) a small chemical group called an acetyl group. Adding acetyl groups (acetylation) can activate (turn on) that part of the chromosome, while taking them away (deacetylation) can deactivate it (turn it off). Drugs called histone deacetylase (HDAC) inhibitors can help in the treatment of some types of cancer by turning on genes that help control cell growth and division.
- RNA interference: Inside each cell, DNA acts as long-term storage for our genes. But DNA isn’t in the same part of the cell where proteins are made. For a protein to be made, a copy of its genetic code (in the form of messenger RNA, or mRNA), needs to be made from the DNA first. This piece of mRNA can then bring the instructions to the part of the cell where proteins are made. mRNA is only used for a short time to make the protein, and then it’s broken down. If the cell needs more of that protein, it makes more mRNA. RNA interference is another way cells can turn off genes. A cell can make other forms of RNA that stick to mRNA. This can cause the mRNA to break down or stop it from delivering its code. Drugs are being developed to target the forms of RNA involved in RNA interference. This might help turn off specific genes that cause cancer.
_
How changes in genes can affect cancer risk:
Some genes normally help control when our cells grow, divide to make new cells, repair mistakes in DNA, or cause cells to die when they’re supposed to. If these genes aren’t working properly, it can affect cancer risk. For example:
- Changes in genes that normally help cells grow, divide, or stay alive can lead to these genes being more active than they should be, causing them to become oncogenes. These genes can result in cells growing out of control.
- Genes that normally help keep cell division under control or cause cells to die at the right time are known as tumor suppressor genes. Changes that turn off these genes can result in cells growing out of control.
- Some genes normally help repair mistakes in a cell’s DNA. Changes that turn off these DNA repair genes can result in the build-up of DNA changes within a cell, which might lead to them growing out of control.
DNA changes that create oncogenes or that turn off tumor suppressor genes or DNA repair genes might lead to cancer, although typically it takes several gene changes before a cell becomes a cancer cell.
Changes in some other genes don’t lead to cancer directly, but they might still make someone more likely to get cancer. For example, some gene changes can limit how well the body breaks down some of the toxins in tobacco smoke. Among people who smoke, people with these kinds of gene changes might be more likely to get lung and other smoking-related cancers.
Gene changes can also play a role in other conditions that might impact cancer risk. For example, some gene variants can affect body weight. People with extra body weight are more likely to get some types of cancer, so these variants might also indirectly affect cancer risk.
_
Normal cell cycle and cell proliferation are regulated by several genes which can be broadly classified into 4 groups viz, proto-oncogenes, tumor suppressor genes, genes regulating apoptosis and genes involved in DNA repair. Some classify genes regulating apoptosis and genes involved in DNA repair as tumor suppressor genes. These genes may be defective due to different factors. The defective genes may lead to production of abnormal proteins which may lead to disruption of the normal cell cycle and proliferation. A single precursor cell with defective gene proliferates surpassing the normal physiologic regulatory process and leads to tumor formation, so, traditionally, it is said that “tumors are clonal”.
Proto-oncogene is a normal gene, which turns defective due to chromosomal rearrangements (e.g., chromosomal inversions, translocations etc), mutations, or gene amplifications and leads to the formation of “oncogenes”. These oncogenes function autonomously and encode defective proteins with erroneous function which affect the cell regulation in a detrimental way. Usually the encoded protein has excessive normal function or acquires a new function. That is why, these mutations are also called as “gain of function” mutations. Similarly, mutations in tumor suppressor genes also alter the normal cell proliferation but these mutations cause “loss of function” of the encoded proteins. The third group of genes which regulate apoptosis, after mutation, causes inhibition of apoptosis leading to increased survival of the cells. The last group of genes which repair any accidental defect in the DNA during cell division may lose their normal function once they are mutated. Mutations can be in the germ line and thus hereditary or they can be acquired due to known ( e.g., environmental factors , viral infections) or unknown causes. In carcinogenesis, most of the mutations are of acquired type.
As it was mentioned, oncogenes (the altered activated protooncogenes) lead to encoding of defective proteins which are known as oncoproteins. These oncoproteins affect the normal cell cycle and enhance the proliferation of the cells. Normally cell proliferation includes a multistep sequential process involving several proteins and interestingly, oncogenes can affect any or several of these steps. In normal physiologic cell proliferation, growth factors bind to growth factor receptors on the cell surface leading to activation of several intracellular signalling pathways in the cytoplasm, which in turn induce and activate regulatory proteins in the nucleus. These activated nuclear factors initiate DNA transcription.
_____
Types of genetic alterations affecting cancer predisposition genes:
Small-Scale Alterations:
At the single-gene level, there are two main types of variants that affect nucleotide sequence: single-base nucleotide variants and small insertions or deletions. Single-base nucleotide variants that are most likely to affect the protein encoded by a gene include missense (change in amino acid), nonsense (introduction of a premature termination codon), loss of the normal initiation codon, loss of the normal stop codon, and alteration of the splice sites in the intronic bases that flank the exons. Occasionally synonymous variants, which do not alter an encoded amino acid, can affect gene function through effects on splicing. Splice sites are located at the boundary of an exon and an intron. Splicing variants in these sequences can disrupt ribonucleic acid (RNA) splicing, resulting in the loss of exons or the inclusion of introns. Insertion or deletions of small nucleotide sequences can result in frameshift variants or in-frame alterations. Frameshift variants almost always lead to a premature termination codon and act similarly to nonsense variants.
Large-Scale Alterations:
Structural variants include deletions, duplications, inversions, and translocations and can occur between chromosomal sequences affecting several exons of the same gene or several genes and bigger chromosomal regions.
_____
_____
Only certain genes can be mutated in ways which make the cells possessing them cancerous. Mutated genes that have this effect are called “cellular oncogenes”, & the normal versions “proto-oncogenes”. Oncogenes were originally discovered because of their introduction into cells by certain oncogenic viruses (i.e. viruses that can convert normal cells into cancerous cells). The hypothesis that viral oncogenes were perturbed versions of normal cellular genes (proto-oncogenes) involved in growth and development that viruses had picked up from cells their ancestors had previously infected opened a new vista on the mechanisms of carcinogenesis (Bishop, 1989). The identification of the sarc protein as the product of the RSV oncogene (v-src) and its cellular counterpart (the product of c-src) confirmed the hypothesis (Brugge and Erikson, 1977; Collett et al., 1978), which was quickly accepted by the scientific community. The 1976 paper by Stehelin, Varmus, and Bishop led to an explosion of work that eventually uncovered a large array of oncogenes that fell into a number of distinct functional categories, depending upon their mechanism of action (Bishop, 1989). The impact of their discovery was reflected in the launch of new journals dedicated solely to oncogenes. Furthermore, it stimulated an intensive investigation of the normal cell cycle, since many oncogenes acted on specific steps in the cycle, resulting in the removal of normal “brakes” on cell proliferation.
_
The very first virus to be discovered to cause cancer was the “Rous sarcoma virus” (RSV); this was discovered about 1910 by Peyton Rous, although he had to overcome so much skepticism that he did not receive the Nobel Prize for this discovery until 1966, when he was very old! This was also the first of the oncogenic viruses that was discovered to contain an extra gene (in addition to the virus’ own genes) responsible for actually causing the transformation. The names given to oncogenes usually consist of 3 letters. For example, the one carried by the Rous sarcoma virus is called by the name src, which is pronounced as if it were “sark”. Other important oncogenes include ras, sis, myc and mos. To distinguish between viral genes and their host equivalents, the viral forms are called “viral oncogenes” (e.g. v-src, v-sis etc.) as opposed to “cellular oncogenes” (c-src, c-sis, c-mos etc.). These normal equivalents are also called “proto-oncogenes”, but this terminology is more appropriate to the situation where the cancer has been caused by somatic mutation, rather than by duplication or over-expression.
_
“Anti-oncogenes” (also called “tumor suppressor genes”) are a special type of genes. Do not be misled by the name to believe either that these genes must be the opposite of oncogenes, or that they must in some way counteract oncogenes. The distinction is more like that between dominant and a recessive mutation, with most oncogenes being dominant, and anti-oncogenes being recessive. In other words, it is the under-activity of the anti-oncogenes that makes the cells possessing them cancerous, rather than over-activity having this result, as is the case with the great majority of oncogenes. As one might suspect from this, both copies of such an anti-oncogene gene (i.e. the copies on both of the two sets of chromosomes) have to be inactivated in order for the cell to become cancerous.
By far the best-studied anti-oncogene is the one called rb (or rb-1), which causes retinoblastoma in humans. This runs in families and results in eventual blindness due to the formation of multiple tumors in both eyes (in the person’s 30s or 40s). Such persons lack a functional rb gene on one of their two sets of chromosomes, so that any retinal cell which undergoes a sufficiently deleterious mutation in the remaining functional copy will give rise to a clone of tumor cells. Another anti-oncogene is called p53, although some mutants of this gene act as if they were ordinary dominant oncogenes, probably because the mutant proteins interact with the normal p53 proteins and prevent their function.
_
Examples of oncogenes:
sis:
Codes for a form of PDGF (Platelet Derived Growth Factor). PDGF normally serves as a cell-to-cell signal, secreted by platelets and diffusing to other cells such as fibroblasts and smooth muscle cells, which it stimulates to grow and crawl about. If a cell produces its own form of PDGF, it behaves as if it were being constantly exposed to high concentrations of external PDGF; in other words, it stimulates its own growth and locomotion without limit. This is called autocrine stimulation. It is thought that the sis protein binds to the PDGF receptor proteins while these receptors are in the cytoplasm (prior to their insertion into the plasma membrane where their normal interaction with PDGF would have occurred).
erbB:
Codes for an abnormal version of the membrane receptor for the extracellular protein called epidermal growth factor. This form of the receptor behaves as if it were constantly bound to molecules of its growth factor, thus constantly sending a false signal stimulating cell growth. A similar oncogene, called erbB-2 seems (based on its base sequence) to code for a receptor for some other (still undiscovered) growth factor. It is found in amplified form in about one fourth of all human breast and ovarian cancers!
ras:
The function of its normal equivalent protein is to relay and amplify stimulatory signals, such as those from growth factor receptors. It binds a molecule of GTP whenever it is itself stimulated. It then relays stimulatory signals and continues to do so until its GTP is hydrolysed to GDP. Certain specific amino acid substitutions eliminate this protein’s ability to hydrolyze bound GTP, however, so that it remains permanently in its “on” state, constantly “relaying” non-existent signals for the cell to grow and divide. Such mutations of this one oncogene are believed to be responsible for no fewer than one fifth of all human cancers, including up to half of colon carcinomas, and 90% of cancers of the pancreas! (Although note that several different ras genes are known.)
src:
Codes for a protein that spontaneously becomes concentrated on the inside surface of the plasma membrane, especially at the sites of cell-substratum adhesion. This protein is an enzyme (a tyrosine kinase) whose effect is to catalyze the covalent bonding of phosphate groups onto the hydroxyl group of tyrosine amino acid residues of proteins. The proteins phosphorylated by the src protein include some which participate in the mechanical linkage between the actin cytoskeleton and materials to which the outside surface of the plasma membrane attaches; these changes may thus be responsible for weakening the cell’s adhesiveness.
myc:
Codes for a nuclear protein whose normal function seems to be as some kind of a transcription factor promoting cell growth. For example, when a normal cell is stimulated to grow and divide (for example, by exposure to PDGF), then the c-myc gene product (protein) temporarily increases in concentration; conversely, this gene normally becomes inactive in non-mitotic cell types. Many human cancers have been shown to have undergone amplification of the c-myc gene (often about 10 copies of the gene) this includes many cases of leukemia and about 30% of lung cancers of the highly lethal “small cell” type and breast cancers. The progression of cancerous cells to ever more and more aggressive states is frequently traceable to further amplification of the myc gene. Most cases of Burkitt’s Lymphoma seem to result from the accidental splicing of one of the myc genes next to the enhancer (control) sequences of the gene for the heavy chain of M antibodies (on the 14th chromosome). Thus, when cells “try to turn on” the genes for making antibodies, the actual result is to turn on transcription of the growth promoting myc genes.
bcl-2:
This name stands for B cell lymphoma and the protein for which this gene codes seems to have the normal function of inhibiting the spontaneous death of B-lymphocytes. In order that the total number of B cells in your body does not continue to increase without limit (because of constant exposure to different antigens) it is essential that the great majority of B-cells self-destruct. When too much bcl-2 protein is produced in a given B cell, this blocks the self-destruction. Trans-genic mice with duplications of the bcl-2 gene accumulate abnormal concentrations of lymphocytes, among other abnormalities. Many human lymphomas result from promoter or enhancer sequences of the antibody genes (on the 14th chromosome) accidentally becoming spliced to the site on the 18th chromosome where the bcl-2 gene is located; whenever these B cells “try” to make antibody molecules, what they make instead is lots of the bcl-2 protein. These cells therefore accumulate to form a slow-growing lymphoma (which is always fatal, although not usually until one of the bcl-transformed clones has subsequently also been transformed by an over-activity of the myc oncogene).
_
Note that most of the oncogenes listed above are part of a “chain of command” by which external signals, especially protein growth factors, normally stimulate cell growth and division. A typical sequence of events would be for a (A) growth factor molecule to diffuse up to a cell’s outer surface, then (B) bind to a membrane protein that serves as a specific receptor for that growth factor, with the conformation (C) or other properties of this receptor being changed by the binding, thereby causing either the activation of a cytoplasmic enzyme (that might be a protein kinase or ), or (D) the activation of a g-protein (such as the c-ras protein) that would then (E) activate a protein kinase, which would phosphorylate various cytoplasmic proteins, including some involved in cell adhesion, and would also stimulate increased transcription and translation of (F) genes for certain transcription factors such as c-myc. Paradoxically, myc also tends to stimulate apoptosis (G), unless counteracted by other gene products such as bcl-2.
_
Cancer cells from actual patients usually contain over-active versions of several different oncogenes, not infrequently 3 or 4 or more. Typically, there is one that acts at the nuclear level (such as myc) and one or more (such as ras or src) that act at the cytoplasmic level. For several years (between about 1982-86) it was confidently believed that cancer wouldn’t occur unless there were at least two, and that one of the two had to be cytoplasmic in its action (like sis, erb, ras or src) and that the other had to act at the nuclear level (like myc). But this is no longer believed.
Assuming that each oncogenic mutation is itself a rather rare event (3 out of 4 people apparently go through their entire lives without having even one such mutation in any of their trillion+ cells!), you should be wondering how more than one of them can arise with any appreciable frequency in a given cell line. The probable answer is that the initial oncogenic change somehow causes an increase in the frequency of subsequent mutations and (in particular) chromosomal breaks and rearrangements. The loss of growth control promotes all sorts of genetic changes, especially chromosomal breaks and the accumulation of extra copies of chromosomes. Most lymphomas are (eventually) found to have many different chromosomal breaks and translocations, sometimes dozens!
_____
_____
Our bodies are made up of trillions of cells, which must work together to keep us healthy. Our cells need to be able to divide to make new cells to help the body grow, or to replace cells that have died. At the same time, cell growth and division need to be controlled, so the cells don’t grow too much and crowd out the cells around them.
It may be helpful to think of a cell as a car. For it to work properly, there need to be ways to control how fast it goes – that is, ways to speed up cell growth and division if it’s needed (like a gas pedal), and ways to keep this growth under control or slow it down (like a brake pedal). There also need to be ways to fix parts of the car if they break down.
Cell growth is normally controlled by the actions of certain genes inside each cell. Cancer begins when cells in the body become abnormal and start to grow out of control. This happens where there are changes in genes that affect cell growth. In order for cells to start dividing uncontrollably, genes that regulate cell growth must be dysregulated. Proto-oncogenes are genes that promote cell growth and mitosis, whereas tumor suppressor genes discourage cell growth, or temporarily halt cell division to carry out DNA repair. Typically, a series of several mutations to these genes is required before a normal cell transforms into a cancer cell. This concept is sometimes termed “oncoevolution.” Mutations to these genes provide the signals for tumor cells to start dividing uncontrollably. But the uncontrolled cell division that characterizes cancer also requires that the dividing cell duplicates all its cellular components to create two daughter cells. The activation of aerobic glycolysis (the Warburg effect), which is not necessarily induced by mutations in proto-oncogenes and tumor suppressor genes, provides most of the building blocks required to duplicate the cellular components of a dividing cell and, therefore, is also essential for carcinogenesis.
The main types of genes that play a role in cancer are:
- Oncogenes
- Tumor suppressor genes
- DNA repair genes (some classify DNA repair genes as subtype of tumor suppressor genes but others say that tumor-suppressor genes act to stop cell growth while DNA repair genes fix errors)
|
Name of Gene |
This Mutation Causes |
Examples |
|
Oncogene |
Uncontrolled growth step on the gas |
Her2-neu, Ras, Myc, Src, Htert |
|
Tumor Suppressor |
Uncontrolled growth: |
P53, Rb, APC |
|
DNA Repair |
No longer able to correct cell |
BRCA1 and BRCA2 |
Cancer is often the result of changes in more than one of these types of genes within a cell.
_
Oncogenes and tumor-suppressor genes exert their effects on tumor growth through their ability to determine cell fates, influence cell survival, and contribute to genome maintenance. The underlying molecular mechanisms can be extremely complex. While tightly regulated in normal cells, oncogenes acquire mutations that typically relieve this control and lead to increased activity of the gene products. This activating mutational event occurs in a single allele. In contrast, the normal function of tumor-suppressor genes is usually to restrain cell growth, and this function is lost in cancer. Because of the diploid nature of mammalian cells, both alleles must be inactivated for a cell to completely lose the function of a tumor suppressor gene. Thus, it requires two genetic events to inactivate a tumor-suppressor gene, while only one genetic event is required to activate an oncogene.
_____
_____
Oncogenes in human cancer:
Work by Peyton Rous in the early 1900s revealed that a chicken sarcoma could be transmitted from animal to animal in cell-free extracts, suggesting that cancer could be induced by an agent acting positively to promote tumor formation. The agent responsible for the transmission of the cancer was a retrovirus (Rous sarcoma virus [RSV]), and the oncogene responsible was identified 75 years later as v-src. Other oncogenes were also discovered through their presence in the genomes of retroviruses that are capable of causing cancers in chickens, mice, and rats. The nonmutated cellular homologues of these viral genes are called proto-oncogenes and are often targets of mutation or aberrant regulation in human cancer. Whereas many oncogenes were discovered on the basis of their presence in retroviruses, other oncogenes, particularly those involved in translocations characteristic of particular leukemias and lymphomas, were identified through genomic approaches. Investigators cloned the sequences surrounding the chromosomal translocations observed cytogenetically and identified the genes activated at the breakpoints. Some of these were oncogenes previously found in retroviruses (like ABL, involved in chronic myeloid leukemia [CML]), whereas others were new (like BCL2, involved in B-cell lymphoma). In the normal cellular environment, proto-oncogenes have crucial roles in cell proliferation and differentiation. Table below is a partial list of oncogenes known to be involved in human cancer.
Oncogenes Commonly Altered in Human Cancers:
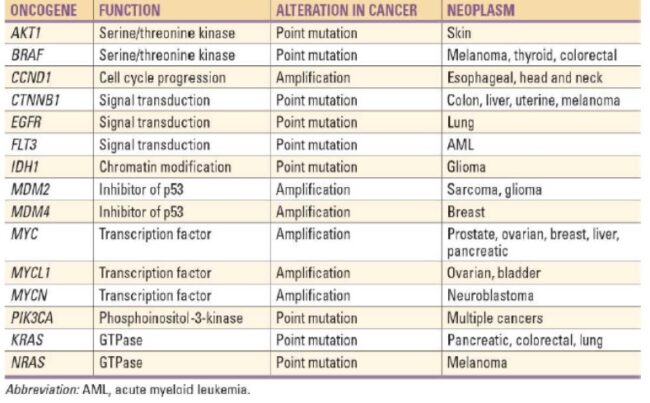
The normal growth and differentiation of cells is controlled by growth factors that bind to receptors on the surface of the cell. The signals generated by the membrane receptors are transmitted inside the cells through signaling cascades involving kinases, G proteins, and other regulatory proteins. Ultimately, these signals affect the activity of transcription factors in the nucleus, which regulate the expression of genes crucial in cell proliferation, cell differentiation, and cell death. Oncogene products function at critical steps in these signaling pathways. Inappropriate activation of these pathways can lead to tumorigenesis.
__
Several different systems have been devised to classify oncogenes. Some of the different categories of oncogenes include:
- Growth factors or mitogens – These oncogenes stimulate the growth, proliferation, and differentiation of cells. Growth factors are usually steroids or proteins secreted by cells to stimulate their own cell proliferation or the proliferation of other nearby or distant cells. These oncogenes can stimulate the cell to secrete growth factors when it would not normally do so, thereby inducing the cell’s abnormal proliferation. An example of an oncogene in this class is c-Sis.
- Receptor tyrosine kinases – Examples of oncogenes in this class include the epidermal growth factor receptor (EGFR), platelet-derived growth factor receptor (PDGFR), vascular endothelial growth factor receptor (VEGFR), and human epidermal growth factor receptor 2 (HER2/neu). The proteins encoded by these oncogenes are very important anti-cancer treatment targets. These kinases act by adding phosphate groups to other proteins to switch them on or off. The receptor kinases add phosphate groups to cell surface receptor proteins that transmit protein signals from the outside to the inside of the cell. Tyrosine kinases add these phosphate groups to tyrosine in the target proteins. This can cause cancer because the receptor is turned on continuously, even in the absence of extracellular signals.
- Cytoplasmic tyrosine kinases – Examples of these oncogenes include the Abl gene in chronic myeloid leukemia (the Philadelphia chromosome) and the Src family, Syk-ZAP-70 family, and BTK family of tyrosine kinases.
- Cytoplasmic serine/threonine kinases – Examples include Raf kinase and cyclin-dependent kinases.
- Regulatory GTPases – An example of a regulatory GTPase is the Ras protein that hydrolyses GTP into GDP and a phosphate. Once activated by a growth factor such as EGF or TGF beta, Ras acts as an on/off switch in major signaling pathways that lead to cellular growth and proliferation.
- Transcription factors – An example is the myc gene, which regulates the transcription of genes that induce cellular proliferation.
______
______
Mechanisms of oncogene activation:
From proto-oncogenes to oncogenes:
Although retroviruses can induce tumour development in animals, only a few instances of human proto-oncogenes’ being mutated into oncogenes by retroviral insertion are known. Nevertheless, various forms of genetic mutation and alteration can convert a human proto-oncogene into an oncogene. Three main mechanisms have been identified: chromosomal translocation, gene amplification, and point mutation.
-1. Point mutation
A proto-oncogene may be converted into an oncogene through a single alteration of a nucleotide. That alteration may be the deletion of a base, the insertion of an extra base, or the substitution of one base for another. Point mutations also can be caused by radiation or chemicals that disrupt the DNA. However, regardless of the type or cause of such a mutation, it usually changes the amino acid sequence of the encoded protein and thus alters protein function. Point mutation (alternatively known as single nucleotide substitution) is a common mechanism of oncogene activation. For example, mutations in KRAS (Kirsten rat sarcoma viral oncogene homolog) are present in >95% of pancreatic cancers and 40% of colon cancers. Activating KRAS mutations are less common in other cancer types, although they can occur at significant frequencies in leukemia, lung, and thyroid cancers. Remarkably— and in contrast to the diversity of mutations found in tumor suppressor genes—most of the activated KRAS alleles contain point mutations in codons 12, 13, or 61. These mutations lead to constitutive activation of the mutant RAS protein. The restricted pattern of mutations observed in oncogenes compared to that of tumor-suppressor genes reflects the fact that gain-of-function mutations must occur at specific sites, while a broad variety of mutations can lead to loss of activity. Indeed, inactivation of a gene can in theory be accomplished through the introduction of a stop codon anywhere in the coding sequence, whereas activations require precise substitutions at residues that can somehow lead to an increase in the activity of the encoded protein under particular circumstances within the cell. A point mutation can increase protein function—as occurs with the ras family of proto-oncogenes—or it can interrupt protein synthesis so that little or no protein is made. Point mutations are common mechanisms of inactivation of tumour suppressor genes.
_
-2. DNA amplification (gene amplification):
The second mechanism for activation of oncogenes is DNA sequence (gene) amplification, leading to overexpression of the gene product. Gene amplification is another type of chromosomal abnormality exhibited by some human tumours. It involves an increase in the number of copies of a proto-oncogene, an aberration that also can result in excessive production of the protein encoded by the proto-oncogene. This increase in DNA copy number may cause cytologically recognizable chromosome alterations referred to as homogeneous staining regions (HSRs) if integrated within chromosomes, or double minutes (dmins) if extrachromosomal. With microarray and DNA sequencing technologies, the entire genome can be surveyed for gains and losses of DNA sequences, thus pinpointing chromosomal regions likely to contain genes important in the development or progression of cancer.
Numerous genes have been reported to be amplified in cancer. Several of these genes, including NMYC and LMYC, were identified through their presence within the amplified DNA sequences of a tumor and their homology to known oncogenes. Because amplified regions often include hundreds of thousands of base pairs, multiple oncogenes may be amplified in a single amplicon in some cancers.
For example, MDM2, GLI1, CDK4, and TPSPAN31 at chromosomal location 12q13-15 have been shown to be co-amplified in several types of sarcomas and other tumors; which of these genes play the causal role in the neoplastic process is still an active area of research. Amplification of a cellular gene is often a predictor of poor prognosis; for example, ERBB2/HER2 and NMYC are often amplified in aggressive breast cancers and neuroblastoma, respectively. The higher the copy number of the N-MYC gene, the more advanced the disease.
__
-3. Chromosomal translocation:
Chromosomal translocation has been linked to several types of human leukemias and lymphomas and, through comprehensive sequencing studies of the genomes of cancers, to epithelial tumours such as prostate cancer. Through chromosomal translocation one segment of a chromosome breaks off and is joined to another chromosome. As a result of such an event, two separate genes can be fused. In some cases the newly created gene leads to tumour development. Such is the case with the so-called Philadelphia chromosome, the first translocation to be linked to a human cancer—chronic myelogenous leukemia. The Philadelphia chromosome is found in more than 90 percent of patients with chronic myelogenous leukemia. This well-known example of translocation involves the fusion of a proto-oncogene called c-ABL, which is located on chromosome 9 encoding a tyrosine kinase, to a site on chromosome 22 known as a breakpoint cluster region (BCR). BCR and the c-ABL gene produce a hybrid oncogene, BCR-ABL, which produces a mutant protein that aberrantly regulates cellular proliferation. The consequence of expression of the BCR-ABL gene product is the activation of signal transduction pathways leading to cell growth independent of normal external signals. Imatinib (marketed as Gleevec), a drug that specifically blocks the activity of Abl tyrosine kinase, has shown remarkable efficacy with little toxicity in patients with CML. The successful targeting of BCR-ABL by imatinib is the paradigm for molecularly targeted anticancer therapies.
Sometimes translocations do not generate a new gene but instead place an intact gene under the control of a regulatory element that normally acts on another gene. That situation occurs in about 75 percent of cases of Burkitt lymphoma. In the cells of patients with this cancer, a proto-oncogene called c-MYC is moved from its site on chromosome 8 to a site on chromosome 14. In its new location the c-MYC gene is positioned next to the switch signal, or promoter region, for the immunoglobulin G gene. As a result, the MYC protein encoded by the c-MYC gene is produced continuously.
Table below lists representative examples of recurring chromosome alterations in malignancy and the associated gene(s) rearranged or deregulated by the chromosomal rearrangement.
Representative Oncogenes at Chromosomal Translocations:
![]()
Translocations are often observed in liquid tumors in general and are particularly common in lymphoid tumors, probably because these cell types have the capability to rearrange their DNA to generate antigen receptors. Indeed, antigen receptor genes are commonly involved in the translocations, implying that an imperfect regulation of receptor gene rearrangement may be involved in their pathogenesis. In addition to transcription factors and signal transduction molecules, translocation may result in the overexpression of cell cycle regulatory proteins or proteins such as cyclins and of proteins that regulate cell death. Recurrent translocations have more recently been identified in solid tumors such as prostate cancers. For example, fusions between TMPRSS2 and ERG, which are normally located in tandem on chromosome 21, contribute to more than one-third of all prostate cancers.
______
Chromosomal instability in solid tumours:
Solid tumors generally contain an abnormal number of chromosomes, a state known as aneuploidy; chromosomes from aneuploid tumors exhibit structural alterations such as translocations, deletions, and amplifications. These abnormalities reflect an underlying defect in cancer cells known as chromosomal instability. While aneuploidy is a striking cellular phenotype, chromosomal instability is manifest as only a small increase in the tendency of cells to gain, lose, or rearrange chromosomes during any given cell cycle. This intrinsically low rate of chromosome aberration implies that cancer cells become aneuploid only after many generations of clonal expansion. The molecular basis of aneuploidy remains incompletely understood. It is widely believed that defects in checkpoints, the quality-control mechanisms that halt the cell cycle if chromosomes are damaged or misaligned, contribute to chromosomal instability. This hypothesis emerged from experimental observations that the tumor suppressor p53 controls checkpoints that regulate the initiation of DNA replication and the onset of mitosis. These processes are therefore defective in many cancer cells. The mitotic spindle checkpoint, which ensures proper chromosome attachment to the mitotic spindle before allowing the sister chromatids to separate, is also altered in some cancers, irrespective of p53 status. The precise relationship between checkpoint deficiency, p53, and chromosomal instability remains unclear, but it is believed that even a subtle perturbation of the highly orchestrated process of cell division can impact the ability of a cell to faithfully replicate and segregate its complement of chromosomes.
In contrast to the genome-wide cytogenetic changes that are typical indications of an underlying chromosomal instability, more focal patterns of chromosomal rearrangement have been recurrently detected in several cancer types. A curious phenomenon known as chromothripsis causes dozens of distinct breakpoints that are localized on one or several chromosomes. These striking structural alterations are thought to reflect a single event in which a chromosome is fragmented and then imprecisely reassembled. While the exact process that underlies chromothripsis remains obscure and its effects on driver genes are not yet clear, a transient period of extreme instability stands in contrast to the gradual loss, gain, and rearrangement of chromosomes that are typically observed in serially cultured cancer cells.
_____
_____
Functions of Oncogene Products:
The viral and cellular oncogenes have defined a large group of genes (about 100 in total) that can contribute to the abnormal behavior of malignant cells. As already noted, many of the proteins encoded by proto-oncogenes regulate normal cell proliferation; in these cases, the elevated expression or activity of the corresponding oncogene proteins drives the uncontrolled proliferation of cancer cells. Other oncogene products contribute to other aspects of the behavior of cancer cells, such as defective differentiation and failure to undergo programmed cell death.
_
Oncoproteins:
Oncoproteins are the product (the proteins) that are coded for by oncogenes and are produced when the gene is transcribed and translated (the process of “writing down the code” on RNA and manufacturing the proteins). There are many types of oncoproteins depending on the specific oncogene present, but most work to stimulate cell growth and division, inhibit cell death (apoptosis), or inhibit cellular differentiation (the process by which cells become unique). These proteins can also play a role in the progression and aggressiveness of a tumor that is already present. The majority of oncogene proteins function as elements of the signaling pathways that regulate cell proliferation and survival in response to growth factor stimulation. These oncogene proteins include polypeptide growth factors, growth factor receptors, elements of intracellular signaling pathways, and transcription factors.
_
There are several subtypes of growth factors, surface receptors, proteins involved in intra-cytoplasmic pathways and intra-nuclear proteins. These processes ultimately lead to progression of a cell in the cell cycle and further cell division. Cell cycles are regulated by cyclins, cyclin dependent kinases and inhibitors. As the above multistep processes involve several subtypes of proteins or factors, mutated protein counterparts or oncoproteins too can be of several subtypes and lead to increased number of mutated cells. Oncoproteins can also affect other processes besides cell cycle, e.g., angiogenesis in tumor, metastasis etc. Some of the oncoproteins, its type and the related cancer have been depicted in Table below:
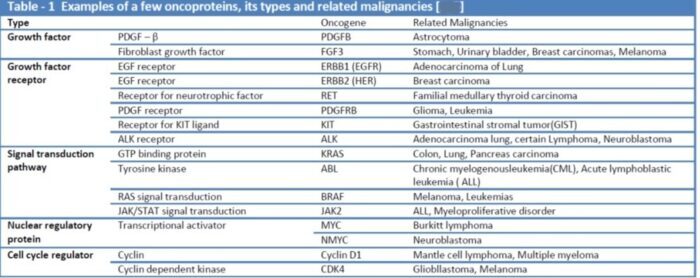
The initial genetic alteration “initiates” the process of tumor formation or carcinogenesis. It was seen in experimental models as well as in some malignancies that tumors may behave differently with time, for example, it may become suddenly aggressive or the response to the treatment regimen may change. It led to the hypothesis that during progression of the tumor, the cells acquire additional genetic alterations which lead to formations of subclones of tumor cells. The traditional concept was that the initial genetic alteration may affect a normal cell which is already differentiated, while the recent concept indicates that stem cell or progenitor cells are the first target. Most hematopoietic tumors and soft tissue sarcomas are initiated by activation of an oncogene, followed by alteration in tumor suppressor genes and other oncogenes. In contrast, most carcinomas are initiated by loss of function of tumor suppressor genes followed by alterations in oncogenes and additional tumor suppressor genes.
_
The action of growth factors as oncogene proteins results from their abnormal expression, leading to a situation in which a tumor cell produces a growth factor to which it also responds. The result is autocrine stimulation of the growth factor-producing cell, which drives abnormal cell proliferation and contributes to the development of a wide variety of human tumors.
_
A large group of oncogenes encode growth factor receptors, most of which are protein-tyrosine kinases. These receptors are frequently converted to oncogene proteins by alterations of their amino-terminal domains, which would normally bind extracellular growth factors. For example, the receptor for platelet-derived growth factor (PDGF) is converted to an oncogene in some human leukemias by a chromosome translocation in which the normal amino terminus of the PDGF receptor is replaced by the amino terminal sequences of a transcription factor called Tel. The Tel sequences of the resulting Tel/PDGFR fusion protein dimerize in the absence of growth factor binding, resulting in constitutive activity of the intracellular kinase domain and unregulated production of a proliferative signal from the oncogene protein. Alternatively, genes that encode some receptor protein-tyrosine kinases, such as erbB-2, are activated by gene amplification. Other oncogenes (including src and abl) encode nonreceptor protein-tyrosine kinases that are constitutively activated by deletions or mutations of regulatory sequences.
_
The Ras proteins play a key role in mitogenic signaling by coupling growth factor receptors to activation of the Raf protein-serine/threonine kinase, which initiates a protein kinase cascade leading to activation of the ERK MAP kinase. As discussed earlier, the mutations that convert ras proto-oncogenes to oncogenes result in constitutive Ras activity, which leads to activation of the MAP kinase pathway. The raf gene can similarly be converted to an oncogene by deletions that result in loss of the amino-terminal regulatory domain of the Raf protein. These deletions result in unregulated activity of the Raf protein kinase, which also leads to constitutive MAP kinase activation.
_
The MAP kinase pathway ultimately leads to the phosphorylation of transcription factors and alterations in gene expression. As might therefore be expected, many oncogenes encode transcriptional regulatory proteins that are normally induced in response to growth factor stimulation. For example, transcription of the fos proto-oncogene is induced as a result of phosphorylation of Elk-1 by the ERK MAP kinase. Fos and the product of another proto-oncogene, Jun, are components of the AP-1 transcription factor, which activates transcription of a number of target genes in growth factor-stimulated cells. Constitutive activity of AP-1, resulting from unregulated expression of either the Fos or Jun oncogene proteins, is sufficient to drive abnormal cell proliferation, leading to cell transformation. The Myc proteins similarly function as transcription factors regulated by mitogenic stimuli, and abnormal expression of myc oncogenes contributes to the development of a variety of human tumors. Other transcription factors are frequently activated as oncogenes by chromosome translocations in human leukemias and lymphomas.
_
G protein-coupled receptors and G proteins also act as oncogenes in some human tumors. For example, mutations of the gene encoding the thyrotropin receptor convert it to an oncogene in thyroid tumors. Thyrotropin is a pituitary hormone that stimulates proliferation of thyroid cells through a G protein-coupled receptor that activates adenylyl cyclase. Mutations of the thyrotropin receptor in thyroid tumors result in constitutive activity of the receptor, which then drives cell proliferation via activation of the cAMP signaling pathway. Likewise, the genes encoding G proteins can act as oncogenes in some cell types. The gsp oncogene, which encodes the α subunit of Gs, is generated by point mutations similar to those found in ras. These mutations result in constitutive activity of the Gs α subunit, leading to unregulated stimulation of adenylyl cyclase. As might be expected, the gsp oncogene is involved in thyroid and pituitary tumors, where cAMP stimulates cell proliferation. Similar mutations convert the genes encoding other G protein α subunits to oncogenes in other cell types, including adrenal and ovarian tumors.
_
The intracellular signaling pathways activated by growth factor stimulation ultimately regulate components of the cell cycle machinery that promote progression through the restriction point in G1. The D-type cyclins are induced in response to growth factor stimulation and play a key role in coupling growth factor signaling to cell cycle progression. Perhaps not surprisingly, the gene encoding cyclin D1 is a proto-oncogene, which can be activated as an oncogene (called D1) by chromosome translocation or gene amplification. These alterations lead to constitutive expression of cyclin D1, which then drives cell proliferation in the absence of normal growth factor stimulation.
_
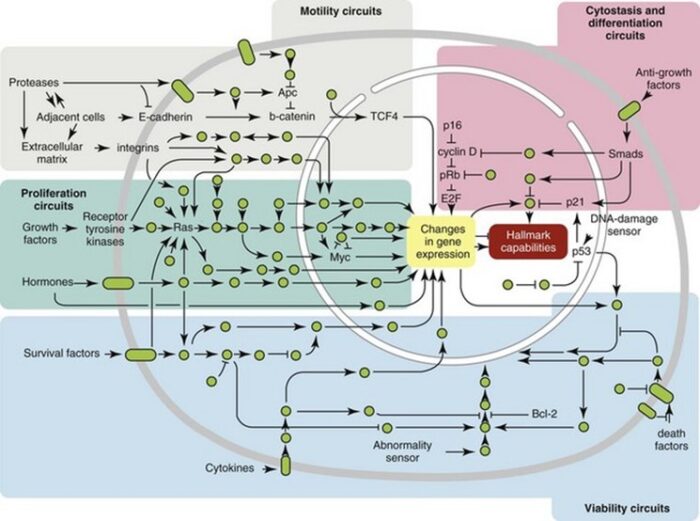
Figure above shows intracellular signaling networks regulate the operations of the cancer cell. An elaborate integrated circuit operates within normal cells and is reprogrammed to regulate hallmark capabilities within cancer cells. Separate subcircuits, depicted here in differently colored fields, are specialized to orchestrate the various capabilities. At one level, this depiction is simplistic because there is considerable crosstalk between such subcircuits. In addition, because each cancer cell is exposed to a complex mixture of signals from its microenvironment, each of these subcircuits is connected with signals originating from other cells in the tumor microenvironment.
_
Although most oncogenes stimulate cell proliferation, the oncogenic activity of some transcription factors instead results from inhibition of cell differentiation. Thyroid hormone and retinoic acid induce differentiation of a variety of cell types. These hormones diffuse through the plasma membrane and bind to intracellular receptors that act as transcriptional regulatory molecules. Mutated forms of both the thyroid hormone receptor (ErbA) and the retinoic acid receptor (PML/RARα) act as oncogene proteins in chicken erythroleukemia and human acute promyelocytic leukemia, respectively. In both cases, the mutated oncogene receptors appear to interfere with the action of their normal homologs, thereby blocking cell differentiation and maintaining the leukemic cells in an actively proliferating state. In the case of acute promyelocytic leukemia, high doses of retinoic acid can overcome the effect of the PML/RARα oncogene protein and induce differentiation of the leukemic cells. This biological observation has a direct clinical correlate: Patients with acute promyelocytic leukemia can be treated effectively by administration of retinoic acid, which induces differentiation and blocks continued cell proliferation.
_
The failure of cancer cells to undergo programmed cell death, or apoptosis, is a critical factor in tumor development, and several oncogenes encode proteins that act to promote cell survival. The survival of most animal cells is dependent on growth factor stimulation, so those oncogenes that encode growth factors, growth factor receptors, and signaling proteins such as Ras act not only to stimulate cell proliferation, but also to prevent cell death. The PI 3-kinase/Akt signaling pathway plays a key role in preventing apoptosis of many growth factor-dependent cells, and the genes encoding PI 3-kinase and Akt act as oncogenes in retroviruses and some human tumors. The downstream targets of PI 3-kinase/Akt signaling include a member of the Bcl-2 family (Bad), and it is notable that Bcl-2 was first discovered as an oncogene in human lymphomas. The bcl-2 oncogene is generated by a chromosome translocation that results in elevated expression of Bcl-2, which blocks apoptosis and maintains cell survival under conditions that normally induce cell death. The identification of bcl-2 as an oncogene not only provided the first demonstration of the significance of programmed cell death in the development of cancer, but also led to the discovery of the role of Bcl-2 and related genes as central regulators of apoptosis in organisms ranging from C. elegans to humans.
_______
_______
Tumour suppressor gene (TSG):
Tumour suppressor genes, like proto-oncogenes, are involved in the normal regulation of cell growth; but unlike proto-oncogenes, which promote cell division and differentiation, tumour suppressors restrain them. If proto-oncogenes are the accelerators of cell growth, tumour suppressor genes are the brakes.
Just as the term oncogene is somewhat misleading because it suggests that the main function of the gene is to cause cancer, the name tumour suppressor gene wrongly suggests that the primary function of those genes is to stem tumour growth. That terminology has to do with the history of their discovery; loss of function of those genes was seen in practically all tumours, and restoration of their function inhibited tumour growth.
Unlike proto-oncogenes, which require that only one copy of the gene be mutated to disrupt gene function, both copies (or alleles) of a particular tumour suppressor gene must be altered to inactivate gene function. In many tumours one copy of a tumour suppressor gene is mutated, producing a gene product that cannot work properly, and the second copy is lost by allelic deletion.
_
Tumor suppressor genes are normal genes that slow down cell division or tell cells to die at the right time (a process known as apoptosis or programmed cell death). When tumor suppressor genes don’t work properly, cells can grow out of control, which can lead to cancer. A tumor suppressor gene is like the brake pedal on a car. It normally helps keep the cell from dividing too quickly, just as a brake keeps a car from going too fast. When something goes wrong with a tumor suppressor gene, such as a pathogenic variant (mutation) that stops it from working, cell division can get out of control. Inherited changes in tumor suppressor genes have been found in some family cancer syndromes. They cause certain types of cancer to run in families. But most tumor suppressor gene mutations are acquired during a person’s lifetime, not inherited. For example, TP53 is an important tumor suppressor gene. It codes for the p53 protein, which helps keep cell division under control. Inherited changes in the TP53 gene can lead to Li-Fraumeni syndrome. Family members with this syndrome have an increased risk of several types of cancer, because all of their cells have this TP53 gene change.
Changes in the TP53 gene are also very common in cancer cells in people without an inherited cancer syndrome. These TP53 changes are acquired during the person’s life. These changes can help the cancer cells grow, but they are found only in the cancer cells, not in other cells in the body, so they can’t be passed on to a person’s children.
_
A tumor suppressor gene (TSG), or anti-oncogene, is a gene that regulates a cell during cell division and replication. If the cell grows uncontrollably, it will result in cancer. When a tumor suppressor gene is mutated, it results in a loss or reduction in its function. In combination with other genetic mutations, this could allow the cell to grow abnormally. The loss of function for these genes may be even more significant in the development of human cancers, compared to the activation of oncogenes. TSGs can be grouped into the following categories: caretaker genes, gatekeeper genes, and more recently landscaper genes. Caretaker genes ensure stability of the genome via DNA repair and subsequently when mutated allow mutations to accumulate. Meanwhile, gatekeeper genes directly regulate cell growth by either inhibiting cell cycle progression or inducing apoptosis. Lastly landscaper genes regulate growth by contributing to the surrounding environment, when mutated can cause an environment that promotes unregulated proliferation. The classification schemes are evolving as medical advances are being made from fields including molecular biology, genetics, and epigenetics.
_
Two-hit hypothesis:
Unlike oncogenes, tumor suppressor genes generally follow the two-hit hypothesis, which states both alleles that code for a particular protein must be affected before an effect is manifested. If only one allele for the gene is damaged, the other can still produce enough of the correct protein to retain the appropriate function. In other words, mutant tumor suppressor alleles are usually recessive, whereas mutant oncogene alleles are typically dominant.
Studies of human hereditary cancers provided compelling evidence for the existence of tumour suppressor genes. In 1971 American researcher Alfred Knudson, Jr., focused on retinoblastoma, which occurs in two forms: a nonhereditary, or sporadic form and a hereditary form that occurs much earlier in life. To explain the differences in tumour rates between those two forms, Knudson proposed a “two-hit hypothesis.” He postulated that in the inherited form of the disease, a child inherits one mutated RB allele from a parent. That single mutation, which is present in every cell, is not sufficient to stimulate tumour formation because the second copy of the RB allele, which is not mutated, functions normally. For a tumour to form, one random mutation must occur in the healthy RB allele of a retinal cell after conception. In contrast, in sporadic cases of retinoblastoma, a sequence of two inactivating events must occur after conception. Because it is much less likely that two random mutation events will occur in the same gene than that one random event will occur, the rate of occurrence of nonhereditary retinoblastoma is much lower than that of the inherited form. Knudson also noted that hereditary cases often developed bilateral tumors and would develop them earlier in life, compared to non-hereditary cases where individuals were only affected by a single tumor.
Knudson’s two hit model has recently been challenged by several investigators. Inactivation of one allele of some tumor suppressor genes is sufficient to cause tumors. This phenomenon is called haploinsufficiency and has been demonstrated by a number of experimental approaches. Tumor-suppressor genes that do not follow the two-hit rule exhibiting haploinsufficiency, includes PTCH in medulloblastoma and NF1 in neurofibroma. Another example is p27, a cell-cycle inhibitor, that when one allele is mutated causes increased carcinogen susceptibility. A certain p53 mutations can function as a dominant negative, meaning that a mutated p53 protein can prevent the function of the natural protein produced from the non-mutated allele.
_
Other tumour suppressor genes that have been discovered through the study of familial cancers include the BRCA1 and BRCA2 genes, which are associated with about 5 percent of hereditary breast cancers; the APC gene, linked to familial adenomatous polyposis coli (a hereditary form of colorectal cancer that causes thousands of polyps to form in the colon, some of which can become cancerous); the WT1 gene, involved in Wilms tumour of the kidney; the VHL gene, associated with kidney cancer and von Hippel-Lindau disease; and the NF1 and NF2 genes, responsible for certain forms of neurofibromatosis.
Tumour suppressor genes discovered through the study of hereditary cancers also play a role in sporadic cancers. For example, hereditary melanoma is associated with a loss of function of the tumour suppressor gene called MTS1 (from multiple tumour suppressor), which also goes awry in a variety of sporadic tumours. MTS1 codes for a protein called p16. When functioning properly, the p16 protein prevents the cell cycle from progressing from the G1 stage to the S stage through an interaction with the RB protein. In cells in which p16 function is lost, the transition from G1 to S is not slowed. That transition point in the cell cycle seems to be extremely important to cellular health, since about 80 percent of human tumours exhibit a problem there.
_
The RB and p53 genes:
Two of the most-studied tumour suppressor genes are RB and p53 (also known as TP53). The RB gene is associated with retinoblastoma, a cancer of the eye that affects 1 in every 20,000 infants. The gene also is associated with bone tumours (osteosarcomas) of children and cancers of the breast, prostate, lung, uterine cervix, and bladder in adults. The p53 gene, which is named for the molecular weight of its protein product (53 kilodaltons), is the most commonly mutated gene in tumours. Practically every person who inherits a mutated copy of a tumour suppressor gene will develop some form of cancer.
_
Loss of function of the RB protein:
The protein E2F is a transcription factor that binds to DNA to stimulate the synthesis of proteins necessary for cell division. When E2F is bound to the RB protein, however, it cannot bind to DNA. Thus, when functioning normally, the RB protein prevents a cell from dividing by binding to E2F. When RB is absent or inactivated, that restraint is lost, and E2F is constantly available to trigger cell division.
_
The p53 gene:
The p53 protein was discovered in 1979. It resides in the nucleus, where it regulates cell proliferation and cell death. In particular, it prevents cells with damaged DNA from dividing or, when damage is too great, promotes apoptosis. Cells exposed to mutagens (chemicals or radiation capable of mutating the DNA) need time to repair any genetic damage they sustain so that they do not copy errors into the DNA of their daughter cells. When mutations occur, normal levels of the p53 protein rise, which slows the transition of the cell cycle from the G1 phase to the S phase. That extra time allows DNA repair mechanisms to effectively restore the DNA sequences to normal. The brakes on the cell cycle—high p53 levels—are then removed, and the cell proceeds to divide.
If there is a large amount of genetic damage, p53 triggers a series of biochemical reactions that cause the cell to self-destruct. Total functional inactivation of the p53 gene will cause genetic damage to accumulate in the cell and will also fail to set off apoptosis in severely injured cells.
Both radiation therapy and chemotherapy can kill tumour cells by stimulating apoptosis. Some tumours that have lost p53 function are more resistant to therapy because of the cells’ diminished capacity to trigger cell death.
Inactivation of the p53 gene occurs through mutation of one allele, and loss of the other accounts for 70 percent of cases of colon carcinoma, 30 to 50 percent of cases of breast cancer, and 50 percent of cases of lung cancer. In two other types of cancer, inactivation of the p53 gene occurs not through mutation and loss of the alleles but through binding of the p53 protein with another protein (called an antagonist) that disables p53 function. One such antagonist, called MDM2, is involved in various tumors. Other antagonists are the “early proteins” produced by cancer-causing strains of the human papillomavirus.
_
Many tumor suppressor genes effect signal transduction pathways that regulate apoptosis, also known as “programmed cell death”. Tumor suppressor genes code for anti-proliferation signals and proteins that suppress mitosis and cell growth. Generally, tumor suppressors are transcription factors that are activated by cellular stress or DNA damage. Often DNA damage will cause the presence of free-floating genetic material as well as other signs, and will trigger enzymes and pathways that lead to the activation of tumor suppressor genes. The functions of such genes is to arrest the progression of the cell cycle in order to carry out DNA repair, preventing mutations from being passed on to daughter cells. The p53 protein, one of the most important studied tumor suppressor genes, is a transcription factor activated by many cellular stressors including hypoxia and ultraviolet radiation damage. Despite nearly half of all cancers possibly involving alterations in p53, its tumor suppressor function is poorly understood. p53 clearly has two functions: one a nuclear role as a transcription factor, and the other a cytoplasmic role in regulating the cell cycle, cell division, and apoptosis as seen in figure below. However, a mutation can damage the tumor suppressor gene itself, or the signal pathway that activates it, “switching it off”. The invariable consequence of this is that DNA repair is hindered or inhibited: DNA damage accumulates without repair, inevitably leading to cancer.
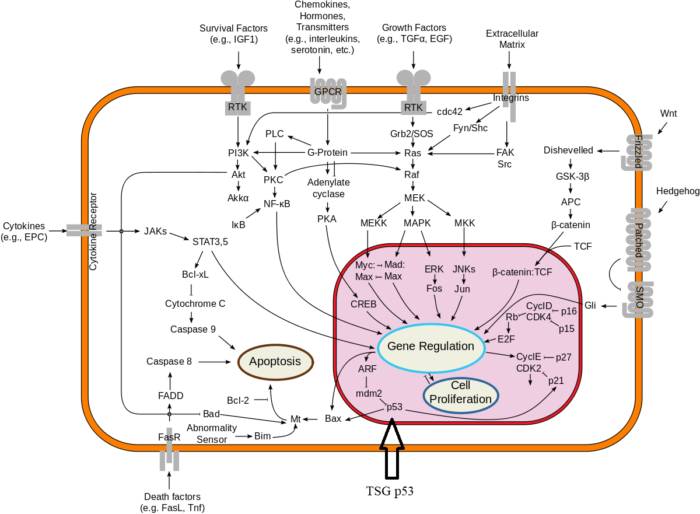
__
Functions of TSG:
The proteins encoded by most tumor suppressor genes inhibit cell proliferation or survival. Inactivation of tumor suppressor genes therefore leads to tumor development by eliminating negative regulatory proteins. In most cases, tumor suppressor proteins inhibit the same cell regulatory pathways that are stimulated by the products of oncogenes. While tumor suppressor genes have the same main function, they have various mechanisms of action, that their transcribed products perform, which include the following:
-1. Intracellular proteins, that control gene expression of a specific stage of the cell cycle. If these genes are not expressed, the cell cycle does not continue, effectively inhibiting cell division. (e.g., pRB and p16)
-2. Receptors or signal transducers for secreted hormones or developmental signals that inhibit cell proliferation (e.g., transforming growth factor (TGF)-β and adenomatous polyposis coli (APC)).
-3. Checkpoint-control proteins that trigger cell cycle arrest in response to DNA damage or chromosomal defects (e.g., breast cancer type 1 susceptibility protein (BRCA1), p16, and p14).
-4. Proteins that induce apoptosis. If damage cannot be repaired, the cell initiates programmed cell death to remove the threat it poses to the organism as a whole. (e.g., p53).
-5. Cell adhesion. Some proteins involved in cell adhesion prevent tumor cells from dispersing, block loss of contact inhibition, and inhibit metastasis. These proteins are known as metastasis suppressors. (e.g., CADM1)
-6. Proteins involved in repairing mistakes in DNA. Caretaker genes encode proteins that function in repairing mutations in the genome, preventing cells from replicating with mutations. Furthermore, increased mutation rate from decreased DNA repair leads to increased inactivation of other tumor suppressors and activation of oncogenes. (e.g., p53 and DNA mismatch repair protein 2 (MSH2)).
-7. Certain genes can also act as tumor suppressors and oncogenes. Dubbed Proto-oncogenes with Tumor suppressor function, these genes act as “double agents” that both positively and negatively regulate transcription. (e.g., NOTCH receptors, TP53 and FAS).
_
Epigenetic influences on TSG:
One of the common and perhaps the most permanent and stable mechanism of epigenetic gene inactivation is the methylation of the 5-carbon of the DNA base cytosine in the 5′-CpG-3′ dinucleotide sequence context of CpG island or promoter regions. These methylation reactions carried out by DNA cytosine methyltransferases are a main component of epigenetic regulatory mechanisms in mammals. In tumor tissues, tumor suppressor genes are often inactivated epigenetically by methylation when compared with normal tissue. Expression of genes, including tumor suppressors, can be altered through biochemical alterations known as DNA methylation. Methylation is an example of epigenetic modifications, which commonly regulate expression in mammalian genes. The addition of a methyl group to either histone tails or directly on DNA causes the nucleosome to pack tightly together restricting the transcription of any genes in this region. This process not only has the capabilities to inhibit gene expression, it can also increase the chance of mutations. Stephen Baylin observed that if promoter regions experience a phenomenon known as hypermethylation, it could result in later transcriptional errors, tumor suppressor gene silencing, protein misfolding, and eventually cancer growth. Baylin et al. found methylation inhibitors known as azacitidine and decitabine. These compounds can actually help prevent cancer growth by inducing re-expression of previously silenced genes, arresting the cell cycle of the tumor cell and forcing it into apoptosis. In addition to DNA methylation, other epigenetic modifications like histone deacetylation or chromatin-binding proteins can prevent DNA polymerase from effectively transcribing desired sequences, such as ones containing tumor suppressor genes.
_____
_____
DNA repair genes:
When a cell divides to make new cells, it needs to make a new copy of all of its DNA. This is a complex process, and sometimes it results in mistakes in the DNA. Genes known as DNA repair genes act like a person who repairs a car. They help fix mistakes in the DNA, or if they can’t fix them, they trigger the cell to die so the mistakes can’t cause any further problems. When something goes wrong with one of these DNA repair genes, it can allow more mistakes to build up inside the cell. Some of these might affect other genes, which could lead to the cell growing out of control. As with other types of gene changes, changes in DNA repair genes can either be inherited from a parent or acquired during a person’s lifetime. Examples of DNA repair genes include the BRCA1 and BRCA2 genes. People who inherit a pathogenic variant (mutation) in one of these genes have a higher risk of some types of cancer, particularly breast and ovarian cancer among women. But changes in these genes are also sometimes seen in tumor cells in people who did not inherit one of these mutations.
_
The primary structure of DNA is constantly subjected to alteration by cellular metabolites and exogenous DNA-damaging agents. These alterations may cause complex genetic changes, including deletions, fusions, translocations and aneuploidy, or, alternatively, single-base changes, which can be either transversions (change of purine to pyrimidine or vice versa) or transitions (change of purine to another purine or pyrimidine to another pyrimidine). Such alterations may ultimately lead to cellular death of unicellular organisms or degenerative changes and aging of multicellular organisms.
_
There are many kinds of DNA lesions occurring in vivo, which are repaired by different DNA repair pathways. These pathways include (i) direct repair of alkyl adducts by O6-alkylguanine DNA alkyltransferase (AGT); (ii) repair of base damage and single-strand breaks (SSBs) by base excision repair (BER); (iii) repair of double-strand breaks (DSBs) by homologous recombination (HR), non-homologous end joining (NHEJ) and single-strand annealing (SSA); (iv) repair of bulky DNA adducts by nucleotide excision repair; (v) repair of cross-links by DNA interstrand cross-link repair and (vi) repair of mismatches and insertion/deletion loops by DNA mismatch repair (MMR).
The critical role played by DNA repair in the maintenance of genome stability is underpinned by the fact that many enzymes involved have been conserved through evolution. Germ line mutations in several of the DNA repair genes are the cause of cancer-predisposing syndromes, and are associated with an increased rate of chromosome breakage and mutagenesis.
_
DNA repair mechanisms are involved in maintaining the integrity of DNA, which often acquires errors during replication. The gene products that oversee the maintenance of DNA integrity help to detect the damage and activate and direct the repair machinery, thereby disabling mutagenic molecules before they permanently damage the DNA. In general, those genes, referred to as the “caretakers of the genome,” behave similarly to tumour suppressor genes. When the cellular mechanisms that repair errors in the DNA are damaged—through acquired or inherited alterations—the rate of genetic mutation increases by several orders of magnitude.
Defects in two mismatch repair genes, called MSH2 and MLH1, underlie one of the most-common syndromes of inherited cancer susceptibility, hereditary nonpolyposis colon cancer. That form of colorectal cancer accounts for 15 to 20 percent of all colon cancer cases. Inherited or acquired alterations in the mismatch repair genes allow mutations—specifically point mutations and changes in the lengths of simple sequence repetitions—to accumulate rapidly (behaviour referred to as a mutator phenotype). Since that defect is inherited by all the cells in the body, it is not known why some organs are more susceptible to cancer development than others.
Another type of repair system that can malfunction is one that corrects defects inflicted on DNA by ultraviolet radiation, a major constituent of sunlight. That kind of radiation damage involves the fusion of two nucleotide bases called pyrimidines to form a “pyrimidine dimer.” Normally, the repair system removes the dimer from the DNA and replaces it with two undamaged nucleotides. Malfunction of the repair pathway, on the other hand, is responsible for two inherited disorders, xeroderma pigmentosum and Cockayne syndrome.
_
BRCA1
Breast cancer type 1 susceptibility protein is a protein that in humans is encoded by the BRCA1 gene. Orthologs are common in other vertebrate species, whereas invertebrate genomes may encode a more distantly related gene. BRCA1 is a human tumor suppressor gene (also known as a caretaker gene) and is responsible for repairing DNA. BRCA1 is also classified as DNA repair gene. The human BRCA1 gene is located on the long (q) arm of chromosome 17 at region 2 band 1, from base pair 41,196,312 to base pair 41,277,500.
BRCA1 and BRCA2 are unrelated proteins, but both are normally expressed in the cells of breast and other tissue, where they help repair damaged DNA, or destroy cells if DNA cannot be repaired. They are involved in the repair of chromosomal damage with an important role in the error-free repair of DNA double-strand breaks. If BRCA1 or BRCA2 itself is damaged by a BRCA mutation, damaged DNA is not repaired properly, and this increases the risk for breast cancer. BRCA1 and BRCA2 have been described as “breast cancer susceptibility genes” and “breast cancer susceptibility proteins”. The predominant allele has a normal, tumor suppressive function whereas high penetrance mutations in these genes cause a loss of tumor suppressive function which correlates with an increased risk of breast cancer.
BRCA1 combines with other tumor suppressors, DNA damage sensors and signal transducers to form a large multi-subunit protein complex known as the BRCA1-associated genome surveillance complex (BASC). The BRCA1 protein associates with RNA polymerase II, and through the C-terminal domain, also interacts with histone deacetylase complexes. Thus, this protein plays a role in transcription, and DNA repair of double-strand DNA breaks ubiquitination, transcriptional regulation as well as other functions.
_
‘Every Hour Hurts, The Last One Kills’. That is an old saying about getting old. Every day, thousands of DNA damaging events take place in each cell of our body, but efficient DNA repair systems have evolved to prevent that. However, our DNA repair system and that of most other organisms are not perfect. In many instances, accumulation of DNA damage has been linked to cancer, and genetic deficiencies in specific DNA repair genes are associated with tumor-prone phenotypes. In addition to mutations, which can be either inherited or somatically acquired, epigenetic silencing of DNA repair genes may promote tumorigenesis.
Effective DNA repair is at the backbone of cancer-free survival. Mutations in DNA repair genes of the nucleotide excision repair (NER) group (XP genes in xeroderma pigmentosum patients), mutations affecting the mismatch repair (MMR) genes [in patients with inherited colorectal cancer (CRC) predisposition], DNA crosslink repair (Fanconi anemia genes), and several others are the cause of inherited cancer syndromes. As an alternative mechanism to genetic mutation, a DNA repair system may be inactivated or decreased in effectiveness by epigenetic gene inactivation mechanisms affecting DNA repair genes.
Human cancers exhibit genomic instability and an increased mutation rate due to underlying defects in DNA repair genes. Hypermethylation of CpG islands in gene promoter regions is an important mechanism of gene inactivation in cancer. Many cellular pathways, including DNA repair, are inactivated by this type of epigenetic lesion, resulting in mutator pathways.
_
Epigenetic inactivation of DNA repair genes:
Two major types of DNA repair exist. The first one repairs DNA damage that arises from external sources such as UV light or ionizing rays and from endogenous DNA damage, for example, due to oxidative stress. To this type of repair belong the base excision repair (BER) pathway, the direct reversal of DNA damage, and the NER pathways. The other general mechanism of repair deals with the mistakes made during DNA replication. This system includes factors involved in MMR, homologous recombination, certain DNA helicases, editing and processing nucleases, and other genes, which are defective in diseases associated with sensitivity to DNA damaging agents (Jackson and Bartek, 2009; Ciccia and Elledge, 2010).
Epigenetic inactivation of DNA repair genes in cancer has been reported for several DNA repair pathways including BER, NER, DNA MMR, and several other DNA damage processing mechanisms. Within one DNA repair pathway, specific genes are often preferentially methylated. It remains to be determined whether this specificity is due to selection of particular repair gene silencing events in promoting tumorigenesis or is due to preferential targeting of the DNA methylation machinery to specific DNA repair gene promoters.
It can be assumed that these epigenetic inactivation processes can result in an increase in genetic instability during tumorigenesis that can be directly attributed to the deficiencies in DNA repair. Therefore, inactivation of DNA repair genes can be seen as an important event in cancer initiation and/or progression by reducing genomic stability leading to genetic aberrations at other important gene loci. Such a mechanism is proven for inactivation of MMR pathways in colorectal tumors but awaits direct confirmation for a number of other DNA repair genes that are found methylated in tumors. On the other hand, diminished DNA repair is expected to lead to reduced cell survival in general, and additional events are likely occurring that enable a cell with reduced repair capacity to undergo uncontrolled proliferation instead of cell death (e.g. mutation in TP53).
_____
_____
Cancer genomes:
The advent of relatively inexpensive technologies for rapid and high throughput DNA sequencing has facilitated the comprehensive analysis of numerous genomes from many types of tumors. This unprecedented view into the genetic nature of cancer has provided remarkable insights. Most cancers do not arise in the context of a mutator phenotype, and accordingly, the number of mutations in even the most advanced cancers is relatively modest. Common solid tumors harbor 30–70 subtle mutations that are nonsynonymous (i.e., result in an amino acid change in the encoded protein). Liquid tumors such as leukemias, as well as pediatric tumors, typically have fewer than 20 mutations. The vast majority of the mutations detected in tumors are not functionally significant; they simply arose by chance in a single cell that gave rise to an expanding clone. Such mutations, which provide no selective advantage to the cell in which they occur, are known as passenger mutations. As noted above, only a small number of the mutations confer a selective growth advantage and thereby promote tumorigenesis. These functional mutations are known as driver mutations, and the genes in which they occur are called driver genes.
_
The frequency and distribution of driver mutations within a single tumor type can be represented as a topographical landscape. The picture that emerges from these studies reveals that most genes that are mutated in tumors are actually mutated at relatively low frequencies, as would be expected of passenger genes, whereas a small number of genes (the driver genes) are mutated in a large proportion of tumors. It appears that there are only a total of ∼120 bona fide driver genes contributing to the development of solid tumors of all kinds, though other driver genes that play a role in a small fraction of cancers are still being discovered. The majority of the mutations in driver genes provide a direct selective growth advantage by altering the signaling pathways that mediate cell survival or the determination of cell fate. The remaining driver gene mutations indirectly provide a selective growth advantage by accelerating the mutation rate of proto-oncogenes and tumor suppressor genes. That the same driver genes play a role in multiple cancer types was unexpected before their discovery and has important implications for the development of new “tumor-agnostic” therapeutic and diagnostic approaches. Moreover, the functions of all these driver genes can be organized into a small number of signaling pathways, as shown in Table below.
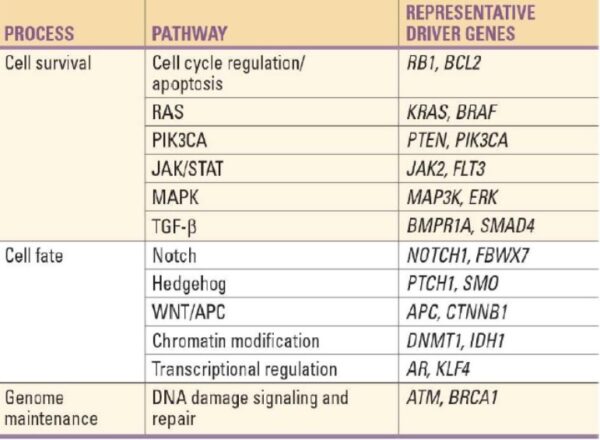
Table above shows signaling pathways altered in Cancer:
_
As a consequence of the mutations they harbor, cancer cells invariably express mutant proteins that are only rarely found in normal cells. Some of these mutant proteins are processed and displayed on the cell surface in the context of major histocompatibility complexes, a process that would normally facilitate their recognition by the adaptive immune system. Thus, all cancers have the theoretical potential to be recognized as foreign, or “nonself,” via the display of these tumor-specific antigens, known as mutation-associated neoantigens (MANAs). In fact, established tumors invariably prevent the activation of local T cells by inducing an intercellular suppressive mechanism known as an immune checkpoint. Therapeutic approaches to exploit this potential vulnerability by blocking immune checkpoints have elicited striking responses in patients with several types of cancer. It was hypothesized that the potential immunogenicity of a tumor would be related to the total number of distinctive neoantigens it can express, which in turn is directly determined by the total number of mutations in the cancer genome. This does seem to be the case. Colorectal cancers that develop as a result of mismatch repair deficiency and smoking-related lung cancers, both of which characteristically harbor large numbers of mutations, exhibit more robust responses to therapeutic immune checkpoint blockade than most other tumor types. Notably, driver mutations as well as passenger mutations that result in the expression of mutant proteins can both contribute to the display of immunogenic neoantigens. Thus, the total number of coding mutations, a metric known as mutational load, is one of the determinants of potential immunogenicity.
_____
_____
Cancer and epigenetics:
Epigenetic events are those that can alter phenotype without changing the genotype. Epigenetics involves genetic control by factors other than an individual’s DNA sequence. Epigenetic changes can switch genes on or off and determine which proteins are transcribed. Epigenetics is involved in many normal cellular processes. Consider the fact that our cells all have the same DNA, but our bodies contain many different types of cells: neurons, liver cells, pancreatic cells, inflammatory cells, and others. How can this be? In short, cells, tissues, and organs differ because they have certain sets of genes that are “turned on” or expressed, as well as other sets that are “turned off” or inhibited. Epigenetic silencing is one way to turn genes off, and it can contribute to differential expression. Silencing might also explain, in part, why genetic twins are not phenotypically identical. In addition, epigenetics is important for X-chromosome inactivation in female mammals, which is necessary so that females do not have twice the number of X-chromosome gene products as males (Egger et al., 2004). Thus, the significance of turning genes off via epigenetic changes is readily apparent. Within cells, there are three systems that can interact with each other to silence genes: DNA methylation, histone modifications, and RNA-associated silencing.
_
During cell division, the DNA with all its modifications is replicated in the S phase and later distributed equally. Even before replication begins, all other cellular components are also replicated. It is generally accepted that their molecular structure not only corresponds to the genomic template but is also altered and modified by a variety of different steps (Alberts et al., 2011). These changes are individual to each cell, and they represent, in aggregate, the adaptations that each individual cell must make in order to function properly in a specific environment (Raj and van Oudenaarden, 2008). They are a result of the cellular environment, the molecules that have been taken up and already processed in a wide variety of ways, the energy available, and the mechanical barriers that allow reactions to take place in a limited space. These countless individual reactions and modifications are significantly influenced and controlled by the local conditions. This has consequences for heredity. If each cell has an individual composition and active cellular working processes (Balázsi et al., 2011; Loewer and Lahav, 2011), the newly synthesized molecules must correspond, in their modifications, to those of the individually adapted existing molecules. This ensures that the genomic, nuclear, and cytosolic composition of the daughter cells is as similar as possible to that of the mother cell and that they are optimally prepared for the existing environmental milieu.
_
Against this background, it is assumed that individual changes will continue to increase over the course of life. This means that in addition to genomic aging (= increasing mutation rate, Yokoyama et al., 2019), a cell also ages metabolically or, if you will, the entity of molecular interaction (EoMI). If we consider the number of substances that humans ingest in the course of their lives (nutrients, salts, water, strong and toxins, inert, lipophilic, hydrophilic substances, etc.) and the stressors to which tissues are exposed (hypoxia, glucose abuse, metabolic disorders, inflammation, infections, radiation, etc.), it becomes clear what external influence cells are subjected to, and to which they then react individually both genetically and with their molecular interactions. These influences lead to changes that permanently manifest themselves genetically, epigenetically, and at the EoMI level, continuously transforming the cell. Depending on the type of substances and the duration of exposure, the change can not only result in cancer cells in certain cases but could also be the cause of chronic diseases. So everything our body ingests and everything that has an external effect on it fundamentally contributes to the change of our cells and our body.
__
Epigenetics refers to all mitotically heritable changes in gene expression and associated phenotypic traits that are not coded in the DNA sequence itself. These changes are mediated by DNA methylation, histone modifications, and non-coding RNAs. Epigenetic changes can be induced by environmental and nutritional factors, and they are involved in a variety of human cancer types and in other chronic disorders. Many cancer risk factors, including ageing, inflammation, tobacco smoking, alcohol consumption, fungal toxins, biological agents, and diet as well as air and water pollution and certain endocrine disrupters, are associated with epigenome dysregulation. Two well-characterized epigenetic mechanisms regulate gene expression. Gene silencing can occur by methylation of CpG residues in promoter regions, as well as by histone deacetylation. Both of these events interfere with the transcriptional machinery and repress gene expression. The effects of global changes in methylation or deacetylation (e.g., by inactivation of DNA methylases or histone deacetylases) remain incompletely understood, but silencing of specific genes by methylation is implicated in numerous cancers of humans and animals. One important observation is that most (or all) genes that are subject to silencing by methylation in specific cancers (e.g., CDKN2A in T-cell leukemia) are commonly inactivated by mutation or deletion in other cancers (e.g., CDKN2A in melanoma).
_
In the past, the term “epigenetics” was used to describe all biological phenomena that do not follow normal genetic principles. Nowadays, epigenetics refers to the study of all changes in gene expression that are transmitted across cell generations and that do not involve changes in the DNA sequence (i.e. mutations).
Consistent with the importance of epigenetic mechanisms in critical cellular processes, dysregulation of epigenetic mechanisms has been linked to various noncommunicable diseases in humans, most notably cancer. Almost all critical processes in cancer cells – such as self-sufficiency in growth signals, insensitivity to anti-growth signals, tissue invasion and metastasis, limitless replicative potential, sustained angiogenesis, and evasion of apoptosis – can be caused not only by genetic changes but also by epigenetic alterations as seen in the figure below:
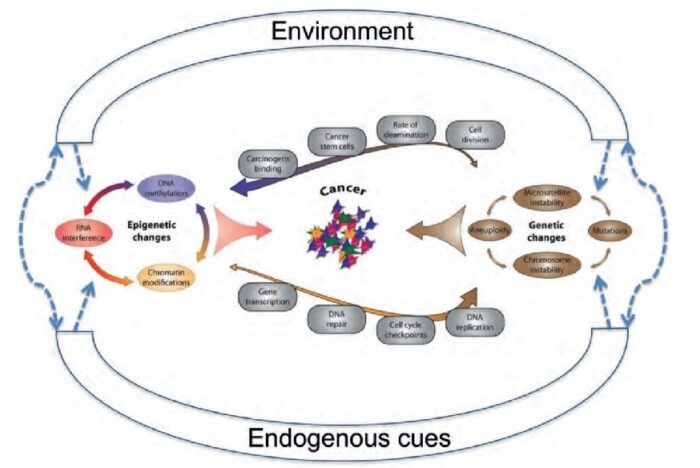
Figure above shows interplay between genetics and epigenetics in cancer development. Epigenetic mechanisms regulate key cellular processes (such as gene transcription, DNA repair, and differentiation) and play critical roles in cellular responses to environmental exposures and endogenous stimuli. Dysregulation of epigenetic mechanisms may promote the development of abnormal phenotypes and cancer. There is cross-talk between epigenetic and genetic changes in the process of cancer development and progression. Given that epigenetic and genetic changes coexist in all cancers, it is important to identify the functionally important changes (“drivers”) that are pertinent to carcinogenesis, and to distinguish them from events that are not functionally important (“passengers”).
It has been proposed that the epigenome may function as an interface between environmental factors and the genome; however, the epigenetic mechanisms by which risk factors induce dysregulation of the epigenome and the functional impact of this dysregulation in specific human cancers remain poorly understood.
_
The inheritance of information based on gene expression levels is known as epigenetics, as opposed to genetics, which refers to information transmitted on the basis of gene sequence. The main epigenetic modification in mammals, and in particular in humans, is the methylation of the cytosine nucleotide residue in CG dinucleotide sequences. Methylation of promoter CpG islands, CG-rich regions that coincide with the promoter of protein-coding genes, correlates with transcriptional silencing. In a normal cell, the DNA methylation patterns are maintained through cell divisions allowing the expression of the particular set of cellular genes necessary for that cell type and blocking the expression of exogenous-inserted sequences. In addition, cells also store epigenetic information in the post-transcriptional modification profile of histones. Different amino acid residues from histones are targets for a variety of modifications, including lysine acetylation, lysine and arginine methylation and serine phosphorylation, with specific functional significance. The hypermethylation of CpG islands in gene promoter regions is associated with specific histone modifications, including dimethylation of histone H3 at lysine 9, deacetylation at this residue, trimethylation of histone H3 at lysine 27 and loss of the transcriptional activating mark H3K4me2 (5,6,7,8). The interconnection between DNA methylation and the presence/absence of specific histone modifications in gene promoter regions suggests that DNA methylation is part of an epigenetic program that leads to transcriptional silencing. In cancer, the epigenetic equilibrium characteristic of a normal cell undergoes dramatic transformations that can be summarized as follows: (i) transcriptional silencing of tumour suppressor genes by CpG island promoter hypermethylation, (ii) global genomic hypomethylation, (iii) loss of imprinting and (iv) epigenetic lack of the repression of exogenous-inserted sequences. Silencing of some tumor suppressor genes in sporadic cancers occurs more frequently by epigenetic methylation than by mutation or deletion. These different mechanisms of gene silencing are not equivalent, as they each result in specific tumor phenotypes. The epigenetic inactivation of tumour suppressor genes by DNA hypermethylation seems to be tumour-type specific and affects all cellular pathways. Examples of genes suffering this aberrant DNA methylation include genes involved in cell cycle (p16INK4a, p15INK4b, Rb, p14ARF), carcinogen metabolism (glutathione s-transferase p1), cell adherence (e-cadherin 1[CDH], CDH13) and apoptosis (death associated protein kinase-1, target of methylation induced silencing-1). One of the most compelling examples of the role of epigenetic gene silencing in the development of human cancer is the inactivation of DNA repair genes by promoter CpG island hypermethylation.
However, we still do not clearly understand why certain CpG islands are hypermethylated in cancer cells while others remain methylation free. We can hypothesize, as has been done in the case of genetic mutations, that a particular gene is preferentially methylated with respect to others in certain tumour types because its inactivation confers a selective clonal advantage. Another possibility that can explain this local hypermethylation is the role played by the environment and nutrition, since the most hypermethylated tumour types are those of the gastrointestinal tract that are more exposed to external carcinogen agents. If we take in consideration the study from Fraga et al. that reports that patterns of epigenetic modifications of MZ twin pairs diverge as they become older, it is not surprising that external factors like smoking habits, physical activity or diet, among others, together with internal factors can influence the hypermethylation status of specific tumour suppressor genes.
_
Furthermore, although epigenetic changes do not alter the sequence of DNA, they can cause mutations. About half of the genes that cause familial or inherited forms of cancer are turned off by methylation. Most of these genes normally suppress tumor formation and help repair DNA, including O6-methylguanine-DNA methyltransferase (MGMT), MLH1 cyclin-dependent kinase inhibitor 2B (CDKN2B), and RASSF1A. Hypermethylation can also lead to instability of microsatellites, which are repeated sequences of DNA. Microsatellites are common in normal individuals, and they usually consist of repeats of the dinucleotide CA. Too much methylation of the promoter of the DNA repair gene MLH1 can make a microsatellite unstable and lengthen or shorten it. Microsatellite instability has been linked to many cancers, including colorectal, endometrial, ovarian, and gastric cancers (Jones & Baylin, 2002).
_
Epigenetic alterations occur frequently in cancers. As an example, one study listed protein coding genes that were frequently altered in their methylation in association with colon cancer. These included 147 hypermethylated and 27 hypomethylated genes. Of the hypermethylated genes, 10 were hypermethylated in 100% of colon cancers and many others were hypermethylated in more than 50% of colon cancers. While epigenetic alterations are found in cancers, the epigenetic alterations in DNA repair genes, causing reduced expression of DNA repair proteins, may be of particular importance. Such alterations may occur early in progression to cancer and are a possible cause of the genetic instability characteristic of cancers. In sporadic cancers, deficiencies in DNA repair are occasionally caused by a mutation in a DNA repair gene but are much more frequently caused by epigenetic alterations that reduce or silence expression of DNA repair genes. Many studies of heavy metal-induced carcinogenesis show that such heavy metals cause a reduction in expression of DNA repair enzymes, some through epigenetic mechanisms. DNA repair inhibition is proposed to be a predominant mechanism in heavy metal-induced carcinogenicity. Cancers usually arise from an assemblage of mutations and epimutations that confer a selective advantage leading to clonal expansion. Mutations, however, may not be as frequent in cancers as epigenetic alterations.
_
As is true for mutations, gene regulation by epigenetic methylation can occur sporadically or it can be heritable. Genomic imprinting presents a unique example in which heritable epigenetic changes influence cancer predisposition. Genomic imprinting refers to a pattern of gene expression that is determined by the parental origin of the gene; in other words, unlike most genes in which both parental alleles are expressed, only one allele (specifically derived from the mother or from the father, depending on the gene) of an imprinted gene is expressed and the other one is permanently repressed. Epigenetic changes in Wilms’ tumor and in heritable colon cancer (among others) alter the expression of the imprinted allele, leading to loss of imprinting that causes overexpression of the insulin growth factor-2 (IGF2) gene.
______
______
Genomic instability and cancer:
Genome stability is a feature of every organism to preserve and faithfully transmit the genetic material from generation to generation or from one somatic cell to another. This includes an error-free replication of genetic material (DNA or RNA) and the repair of replication mistakes or damaged DNA/RNA. In contrast, genome instability generally refers to the appearance of a high frequency of mutations in a genome, including point mutations, insertion/deletions, and large genetic rearrangements. Genomic instability is a characteristic of most cancer cells. It is an increased tendency of genome alteration during cell division. Genomic instability is a hallmark of cancer development and progression, and characterizing the stresses that create and the mechanisms by which cells respond to genomic perturbations is essential. Cancers are known to exhibit genome instability or a “mutator phenotype”. The protein-coding DNA within the nucleus is about 1.5% of the total genomic DNA. Within this protein-coding DNA (called the exome), an average cancer of the breast or colon can have about 60 to 70 protein altering mutations, of which about 3 or 4 may be “driver” mutations, and the remaining ones may be “passenger” mutations. However, the average number of DNA sequence mutations in the entire genome (including non-protein-coding regions) within a breast cancer tissue sample is about 20,000. In an average melanoma tissue sample (melanomas have a higher exome mutation frequency) the total number of DNA sequence mutations is about 80,000. These high frequencies of mutations in the total nucleotide sequences within cancers suggest that often an early alteration in the field defect giving rise to a cancer is a deficiency in DNA repair. Large field defects surrounding colon cancers (extending to about 10 cm on each side of a cancer) are found to frequently have epigenetic defects in two or three DNA repair proteins (ERCC1, ERCC4 (XPF) and/or PMS2) in the entire area of the field defect. When expression of DNA repair genes is reduced, DNA damage accumulates in cells at a higher than normal rate, and this excess damage causes an increased frequency of mutation and/or epimutation. Mutation rates strongly increase in cells defective in DNA mismatch repair or in homologous recombinational repair (HRR). A deficiency in DNA repair, itself, can allow DNA damage to accumulate, and error-prone translesion synthesis of some of the damaged areas may give rise to mutations. In addition, faulty repair of this accumulated DNA damage may give rise to epimutations. These new mutations and/or epimutations may provide a proliferative advantage, generating a field defect. Although the mutations/epimutations in DNA repair genes do not, themselves, confer a selective advantage, they may be carried along as passengers in cells when the cell acquires an additional mutation/epimutation that does provide a proliferative advantage.
_____
Genomic instability can initiate cancer, augment progression, and influence the overall prognosis of the affected patient. Genomic instability arises from many different pathways, such as telomere damage, centrosome amplification, epigenetic modifications, and DNA damage from endogenous and exogenous sources, and can be perpetuating, or limiting, through the induction of mutations or aneuploidy, both enabling and catastrophic.
_
- • Cellular mechanisms that prevent or promote genomic instability:
Genomic instability plays critical roles in both cancer initiation and progression. This instability can manifest itself genetically on several different levels, ranging from simple deoxyribonucleic acid (DNA) sequence changes to structural and numerical abnormalities at the chromosomal level. Paragraphs below briefly outline the mechanisms that maintain the stability of nuclear and mitochondrial DNA and how these mechanisms may become corrupted in cancer cells.
_
-1. Telomeres foster chromosomal stability and can inhibit or promote malignant transformation:

The chromosome stabilizing role of intact telomeres was recognized as early as the 1930s from independent research by McClintock and Muller and more recent work has further strengthened the connection between telomere dysfunction and chromosomal instability (CIN). Telomeres, which are located at the ends of each chromosome, consist of approximately 5–10 kbp of specialized, tandem repeat, noncoding DNA complexed with a variety of telomere associated proteins. These elements create a protective cap that prevents the recognition of the chromosomal termini as DNA double strand breaks (DSBs) and their consequent aberrant repair via nonhomologous end joining (NHEJ) or homologous recombination (HR). Due to the inability of DNA polymerase to fully replicate the ends of linear DNA molecules, in the absence of compensatory mechanisms, telomeric DNA is lost at the rate of approximately 100 base pairs (bp) per telomere per cell division. In normal somatic cells, this telomere erosion is used by the cell to monitor its division history, with moderate telomere shortening triggering either irreversible cell cycle arrest, termed replicative senescence, or apoptosis. This block to continued proliferation is thought to have evolved to prevent the development of cancer in long-lived organisms by restricting the uncontrolled outgrowth of transformed cell clones, and also by preventing further telomere erosion which would accompany such abnormal growth and eventually destabilize the telomeres leading to CIN.
_
A current popular model for the involvement of telomere shortening in carcinogenesis posits that increasing numbers of cells experience telomere shortening as a person ages, which increases the pool size of cells that are in danger of experiencing eventual telomere dysfunction and prooncogenic CIN. In the vast majority of such cells, the senescence and apoptotic blocks are strictly enforced. However, this process eventually fails in rare cells which continue to replicate and eventually experience CIN due to critical telomere shortening. Notably, such cells may be more tolerant of rampant genomic instability due to their previous abrogation of the tumor suppressive telomere length checkpoints. However, if left unchecked, this instability will eventually reach lethal levels in the transforming cells, thereby presenting a second block to the development of cancer. This escalating telomere driven CIN creates a strong selective pressure for telomere maintenance in incipient cancer cell populations; a problem that is solved in one of two ways: activation of telomerase or alternative lengthening of telomeres (ALT). In the majority of human cancers, the telomere specific reverse transcriptase telomerase, which is stringently repressed in normal somatic cells, is activated, thereby restabilizing the telomeres, although cancer telomeres on average seem to remain very short. Whereas most cancers use telomerase to maintain telomere length, a significant minority of cancers (typically non-carcinomas) utilize ALT, a telomerase independent, homologous recombination based mechanism. This mode of telomere maintenance results in extreme telomere length heterogeneity and, interestingly, better patient survival compared to their telomerase positive counterparts in several tumor types. These observations suggest that cancer cells utilizing ALT may have compromised their vitality in exchange for the unlimited replicative potential conferred by this telomere maintenance mechanism.
_
-2. Centrosomes, the spindle assembly checkpoint, and tumorigenesis:
The centrosome is the primary microtubule organizing center in dividing mammalian cells and is composed of a pair of centrioles surrounded by a cloud of proteins that promote microtubule nucleation. The centrosome is duplicated in a semiconservative fashion with one daughter centriole formed next to a preexisting mother centriole, and this process only occurs once in every cell cycle. Centrosome amplification, the presence of greater than two centrosomes during mitosis, is a common characteristic of most solid and hematological tumors that may induce multipolar mitoses, chromosome missegregation, and subsequent genetic imbalances that promote tumorigenesis.
_
Centrosome amplification may be caused by diverse mechanisms, including centrosome overduplication, de novo assembly, and mitotic failure downstream from mono- or multipolar division. The end result of these structural abnormalities is often cytokinesis failure, which can give rise to tetraploid binucleated cells and genome instability downstream. Over time, the net result is a small population of cells that harbor the ability to manage extra centrosomes, which could account for the accumulation of cancer cells with centrosome amplification and aneuploidy.
_
Catastrophic aneuploidy and nonviable daughter cells are a possible tumor suppressive consequence for centrosome abnormalities. However, cancer cells have developed mechanisms that overcome this fate by clustering multiple centrosomes into a “pseudobipolar” state. Cancer cells may utilize this mechanism to dampen high level aneuploidy and extreme CIN, leading to better prognostic outcomes. Centrosome clustering in tumor cells is not completely understood, but it is likely to rely on microtubule associated proteins and motor proteins that bundle together microtubules and centrosome. Given that centrosome clustering may be advantageous for cancer cell survival, this process may be an attractive and specific therapeutic target. In theory, the induction of multipolarity through declustering of supernumerary centrosomes will selectively target cancer cells without affecting healthy cells.
_
Bipolar chromosome attachment during mitosis is ensured by a quality control mechanism known as the spindle assembly checkpoint. The assembly checkpoint senses tension across kinetochores as a measure of bipolar attachment of chromosomes, and prevents the onset of anaphase in the presence of unattached and/or misattached chromosomes. Any failures to sense errors will compromise the checkpoint and, potentially, induce instability. The assembly checkpoint relies upon kinase signaling to delay cell cycle progression and correct attachment errors. Aurora kinase B, for example, detects misattached chromosomes and overexpression of the kinase is sufficient to disrupt the checkpoint and promote tetraploidy. Moreover, mutations or expression changes in other checkpoint gene products may compromise the checkpoint and favor tumorigenesis. Lastly, oncogenic cues, such as overexpression of Aurora kinase A, may override a functioning checkpoint and enable cells to enter anaphase despite misattached chromosomes. Cancer cells may take advantage of the checkpoint for their own benefit. For example, checkpoint mediated delay provides time for centrosome clustering which can be manipulated by disabling or restoring assembly checkpoint function. The ability to manipulate or hijack the cell’s innate quality control mechanism may act as a selection pressure, and cancer cells that possess this ability may have a growth advantage over others.
_
Correlation between aberrant centrosome numbers and aberrant chromosome numbers dates back over 100 years, yet there is still a debate whether supernumerary centrosomes are the cause or the result of genomic instability, or vice versa. One interesting phenomenon that may shed light on this debate is the presence of a transient tetraploid state during tumorigenesis. Tetraploidy arises after cytokinesis failure following prolonged activation of the assembly checkpoint, regardless of the reason for checkpoint activation. Depending on the status of tumor protein 53 (TP53), a tumor suppressor, the aborted postmitotic cells will either undergo apoptosis after prolonged cell cycle arrest or continue to cycle. In p53 null cells, a postmitotic checkpoint is compromised, which enables the cell to progress through a subsequent cell cycle with double the amount of centrosomes and genetic material. Consequently, each subsequent division for these tetraploid cells will be more error prone, generating more unstable and detrimental aneuploidy. A TP53-dependent postmitotic checkpoint is frequently mutated during early stages of tumorigenesis, which suggests that the tetraploid state serves as an intermediate for the aneuploid state observed in cancer cells. In patients with Barrett’s oesophagus, the presence of tetraploid cells is detected before aneuploid cells and correlates with early loss of TP53. Tetraploid cells were also isolated from p53-/− mouse mammary epithelial cells, and these cells formed tumors in nude mice and showed increased aberrant mitoses and genomic instability in culture. Therefore, regardless of how centrosome amplification or genomic instability occurs in this “chicken or egg” argument, it is clear that either event is positively enhanced by the other in promoting tumorigenesis.
_
-3. Epigenetic mechanisms contributing to genomic instability:
Epigenetics refer to all heritable changes that may modify gene expression without changing the primary DNA sequence, such as DNA methylation and chromatin remodelling. Epigenetic modifications are established during differentiation and are stably inherited and maintained through multiple rounds of cell division. Epigenetic processes that lead to genomic instability and ultimately malignant transformation constitute heritable changes that modulate gene expression and can also affect DNA repair dynamics.
_
DNA methylation consists of the addition of a methyl group at the carbon 5 position of the cytosine pyrimidine ring or the number 6 nitrogen of the adenine purine ring. Most cytosine methylation occurs in the context of cytosine-phosphate-guanine (CpG) dinucleotides, and occurs via a group of DNA methyltransferase enzymes resulting in silencing of gene transcription. Aberrant changes in DNA methylation were among the first events to be recognized in cancer. Global hypomethylation in repetitive sequences of the genome can occur early during tumorigenesis and may initially predispose premalignant cells to repetitive sequence genomic instability. Furthermore, hypomethylation of the promoter of oncogenes can increase their expression and lead to genomic instability. Similarly, aberrant sequence specific hypermethylation in cancer cells can lead to further genomic instability by the silencing of genes involved in cell cycle regulation and DNA repair. A prominent example is the aberrant methylation of CpG islands in the promoter regions of DNA mismatch repair (MMR) genes that result in cancer cells with a “mutator phenotype”.
_
In addition to DNA methylation, histone molecules that form the primary protein component of chromatin also regulate genome stability as well as gene transcription. A number of posttranslational modifications such as acetylation, deacetylation, methylation, phosphorylation and ubiquitination have been identified that alter the function of histones. Various combinations of these posttranslational histone modifications have been hypothesized to form a “histone code” that dictate distinct chromatin structures that can affect genome stability pathways and transcription. Acetylation of the lysine residues at the amino (N) terminus of histone proteins removes positive charges, thereby reducing the affinity between histones and DNA to facilitate access by ribonucleic acid (RNA) polymerase and transcription factors to gene promoter regions. Therefore, in most cases, histone acetylation enhances transcription while histone deacetylation represses transcription. In addition, histone acetylation can affect DNA repair by promoting histone dynamics that stimulate a DNA damage response in response to ionizing radiation. Similarly, histone ubiquitination can also modify DNA repair capacity. Briefly, ubiquitinated histones can lead to chromatin structures that are conducive to the assembly of nucleotide excision repair complexes on damaged DNA, as well as both types of DSB repair pathways and cell cycle checkpoint factors critical for the DNA damage response. Monoubiquitination of histones H2A and H2B prevents chromatin compaction and facilitates assembly of the repair machinery at the damaged sites. Polyubiquitination of histone H2A and H2AX is important for the retention of repair proteins, such as TP53 binding protein 1 (53BP1) and breast cancer 1 (BRCA1), at damaged loci. Finally, histone phosphorylation is an early event following DNA damage and required for efficient DNA repair. Upon introduction of a DSB, hundreds of histone molecules become phosphorylated within minutes at the chromatin flanking the break site, thus providing a rapid and highly amplified detection system and a focus for the accumulation of many other proteins involved in DNA repair and chromatin remodelling. These examples, and numerous other observations, suggest that a vast array of epigenetic mechanisms contribute to the genomic instability in cancer cells.
_
-4. Mitochondrial DNA alteration in human cancers:
Mitochondrial genetic reprogramming and energy balance within cancer cells play a pivotal role in tumorigenesis and are duly regarded as one of the hallmarks of human cancer. In 1927, Otto Warburg, identified mitochondrial dysfunction as a key component of tumorigenesis and numerous studies have since elaborated upon the role of mitochondrial DNA (mtDNA) alterations in different human cancers. Mitochondria are the key component of the oxidative phosphorylation system to generate cellular adenosine triphosphate. They uniquely possess their own DNA and generate reactive oxygen species (ROS). Most human cells contain hundreds of nearly homoplasmic (identical) copies of mtDNA, which are maternally inherited. Compared to the nuclear DNA, the mutation rate of mtDNA is nearly 10 times higher and alterations are much easier to detect due to their high copy number in cancer cells.
_
A substantial number of studies identified somatic mtDNA mutations involving coding and noncoding mtDNA regions in various cancers. Among the noncoding mtDNA mutations, a poly C mononucleotide repeat (known as D310) was frequently altered in numerous cancers and appeared to be a mutational hot spot. Notably, coding mtDNA mutations targeting respiratory complex I, III, IV or V were frequent in a variety of human cancers. Moreover, alteration of mtDNA copy number could potentially be associated with mitochondrial dysfunction leading to disease progression. In recent studies, a correlation between mutations in mtDNA and epidermal growth factor receptor, or prostate-specific antigen expression, was established in lung and prostate cancer, respectively. These results suggest cross talk between mitochondria and nuclear genomes maintain tumor growth.
_
In order to understand their role, a number of studies introduced mtDNA mutations in cancer cells. Introduction of a mitochondrial mutant adenosine triphosphate 6 (ATP6) (complex V) or cyclooxygenase 1 (COXI) (complex IV) increased the growth of prostate cancer cells or induced cancer cell proliferation and altered reactive oxygen and nitrogen species, respectively. In a bladder cancer study, introduction of a mutant mitochondria encoded cytochrome B (CYTB) induced bladder cancer growth and invasion, accompanied with increased ROS, lactate production and oxygen consumption. Moreover, the ROS-producing CYTB mutant tumor cells efficiently killed normal splenic immune effector cells, which may provide tumor cells with an immune evasion mechanism. In addition, mutant CYTB overexpression in nontumorigenic bladder epithelial cells triggered an increased mitochondrial proliferation and inhibition of apoptosis. As these mutations in mtDNA were detected in human patients, the preceding studies suggest a causative role for mtDNA alterations in tumorigenesis.
______
- • Repair pathways responsible for genetic fidelity and tumor suppression:
DNA is replicated with extreme fidelity in normal cells with a mutation rate of about 10-10 per base pair per cell division. DNA damage typically occurs through the following: (1) exposure to agents such as ultraviolet irradiation, genotoxic chemicals, and ionizing radiation; (2) spontaneous DNA damaging events, such as a basic site formation; and (3) failure in normal cellular DNA processing and replication events, such as stalled replication forks. These processes induce oxidation, alkylation, crosslinking, dimerization, and strand breaks in DNA, which must be resolved. As such, repair of this DNA damage is essential to preserving genome integrity and preventing cancer.
_
-1. Excision repair pathways:
Three excision repair pathways can repair single stranded DNA damage: nucleotide excision repair (NER), base excision repair (BER), and DNA mismatch repair (MMR).
Nucleotide excision repair:
Fidelity of genetic information transmission depends on NER, which serves to repair DNA damage caused by ultraviolet irradiation, alkylating and oxidizing agents, or chemotherapeutic drugs that form bulky, helix distorting adducts. Two sub-pathways have been identified. Global genome NER repairs damage in both strands of the DNA regardless of whether the gene is being actively transcribed. Transcriptionally coupled NER, however, repairs transcriptionally active genes. The two pathways are similar in that they use many of the same pathways, but global genome NER uses xeroderma pigmentosum complementation group C (XPC)-RAD23 homolog B (HR23B) and DNA damage binding protein 1 (DDB1)-DDB2/XPE proteins to recognize distortions in the double helix while transcriptionally coupled NER occurs at regions where RNA Polymerase II has stalled. Genetic polymorphisms of NER gene products associate with human diseases, including xeroderma pigmentosum, which can lead to severe cases of skin cancer.
Base excision repair:
The BER pathway fixes damaged DNA bases. These lesions are recognized and removed by specific DNA glycosylases, which cleave the glycosidic bond between the damaged base and the sugar of the DNA backbone. In more complex lesions, proliferating cell nuclear antigen (PCNA), flap endonuclease 1 (FEN1), and DNA polymerase (POL) β, with or without POLδ/ɛ, act to repair the lesion. This complex set of events in BER is facilitated by poly (ADP-ribose) polymerase 1 (PARP1), which recruits proteins involved in the DNA repair response, such as X-ray repair cross-complementing protein (XRCC)1, DNA ligase, and DNA polymerase. Because cells are constantly subjected to DNA damaging conditions, the BER pathway is crucial to preserving genome integrity. This is exemplified by the embryonic lethality of mice that possess knockouts of key components of this pathway. A biallelic germline defect within a DNA glycosylase, mutY Homology (MUTYH), was initially found in families that had excess colorectal tumors with somatic mutations in the adenomatous polyposis coli gene. A subsequent larger study revealed that biallelic germline MUTYH defects conferred 93 fold excess risk of colon cancer with penetrance by age 60 and may also confer increased risk for endometrial cancer. Mutations in another glycosylase, 8 Oxoguanine (OGG1), have been associated with laryngeal cancers while gastric cancers harbor inactivating mutations in glycosylase nei endonuclease VIII-like 1 (NEIL1). Taken together, these studies confirm the importance of BER in the suppression of carcinogenesis.
DNA mismatch repair:
Some evidence suggests that proofreading activity of replicative DNA polymerases and MMR machinery act in series in mammalian cells. MMR targets could generally be classified into base/base mispairs and large insertion–deletion loops. At the forefront of error recognition, MutS protein homolog (Msh) 2 pairs with Msh6 or Msh3, to form MutSα (Msh2/Msh6) and MutSβ (Msh2/Msh3). Whereas the former is mostly responsible for base/base mispairs, the latter targets large insertion–deletion loops. To initiate the repair process, MutL homolog 1(Mlh1)/Pms2 heterodimers (MutL homologues), in the presence of exonuclease1, interact with MutS complexes to create nicks in the 3′ and 5′ of the nascent strand containing the mismatch. Following nick creation, enzymes required to repair the damage site are recruited and resynthesis of the DNA is carried out by POLδ/ɛ. As a nexus for DNA damage sensing and cell death, MMR machinery play an important role in recognizing damaged DNA and relaying signals downstream to activate a G2/M cell cycle checkpoint.
_
-2. Double strand break repair and cancer predisposition:
The DSB is the most lethal form of DNA damage, as it can lead to significant DNA damage by multiple genomic changes, including translocation formation, deletions, and amplifications, resulting in heritable cellular genomic instability/damage that can lead to malignancy. DSBs are repaired by both HR and NHEJ repair pathways. NHEJ repair occurs throughout the cell cycle, while HR prevails in S and G2 phase cells. NHEJ repair joins broken DNA ends without identifying DNA sequence homology and is therefore highly error prone. HR repair is dependent upon DNA sequence homology and therefore is relatively error free.
_
Errors in the NHEJ pathway may generate inappropriate dicentric chromosomes that are covalently joined. These dicentric chromosomes may break during anaphase, producing new dicentric chromosomes through further NHEJ. This process is known as the breakage fusion bridge (BFB) cycle, which is important in telomere related genome instability. Damaged telomeres will be processed by NHEJ, unless the broken ends are healed by a new telomere, and continuation of the BFB cycle can result in complex chromosomal rearrangements that include gene loss, gene amplification and unbalanced translocations. BFB cycles are self-perpetuating and result in genetic heterogeneity in a variety of cancers.
_
HR repair involves multiple gene products some of which are involved in repair of stalled replication forks. The DSB is recognized by a Mre11-Rad50-NBS1 complex which recruits many different proteins, including proteins with topoisomerase, endonuclease, and helicase activity. Eventually a “synaptic complex” is formed which allows homologous single strand DNA (ssDNA) to invade and anneal to complementary DNA. DNA polymerase then fills in the ssDNA gap and the synaptic complex is resolved. Both crossover and non-crossover products can be created by this process. Interestingly, loss or mutation of many of these gene products is associated with specific cancer prone diseases.
_
BRCA1 and BRCA2 are key players in the HR repair pathway and act as tumor suppressors by maintaining genome stability. Linkage studies in families with early onset breast cancer detected the presence of a breast cancer susceptibility gene, BRCA1. Subsequently, mutations in BRCA1 were confirmed in families with early onset breast and ovarian cancer. Later, a novel locus, encoding BRCA2, was discovered and linked to breast cancer susceptibility. In one meta-analysis, cumulative breast and ovarian cancer risks for BRCA1 mutation carriers are 57 and 40%, respectively, while, for carriers of BRCA2 mutations, these risks are 49 and 18%, respectively. Furthermore, hereditary BRCA mutations have been also linked to pancreatic, prostate, and colon cancers. Of interest, germline mutations in BRCA1 versus BRCA2 associate with different subtypes of breast cancer. BRCA1 associated cancers are of the more aggressive triple negative subtype and appear at an earlier age than sporadic tumors. In contrast, BRCA2-associated tumors relate mostly with hormone receptor positive breast cancers.
_
Like BRCA1 and BRCA2, partner and localizer of BRCA1 (PALB2) promotes genome integrity through its role in DSB repair. It binds and colocalizes with BRCA2 in the nucleus to stabilize BRCA2 foci and facilitate the intra S phase checkpoint and HR repair. Germline mutations in PALB2 confer a 2–5 fold increase in breast cancer risk and germline mutations have been recently found in African American breast cancer patients. PALB2 mutations have also been observed in 1% of Chinese women with early onset breast cancer. Interestingly, exome sequencing identified PALB2 as a pancreatic cancer susceptibility gene and PALB2 mutations have been found in patients with familial pancreatic cancers.
_
Fanconi anemia (FA) is an autosomal recessive disorder characterized by congenital defects, CIN, hypersensitivity to DNA crosslinks, bone marrow failure, and predisposition to cancer. Fifteen FA or FA-like genes have been identified, all of which are involved in coordinating DNA repair through the FA/BRCA pathway. Interestingly, two of these genes are BRCA2 (FANCD1) and PALB2 (FANCN), thus revealing interplay between FA and HR. Patients with FA have increased susceptibility to breast, ovarian, and oral cancers. Additionally, heterozygote carriers of germline mutations in FA genes may also harbor an increased risk to develop cancer. Importantly, unlike the BRCA-associated cancers, tumors from FA patients, or with acquired FA defects, may be hypersensitive to crosslinking agents such as cisplatin and mitomycin C, and also are hypersensitive to radiation. Finally, biallelic loss of ataxia telangiectasia mutated (ATM) results in ataxia telangiectasia, a disease characterized by a roughly 1000 fold increased lymphoma incidence.
______
______
Mitotic catastrophe: a mechanism for avoiding genomic instability, a 2011 study:
The improper distribution of chromosomes during mitosis compromises cellular functions and can reduce cellular fitness or contribute to malignant transformation. As a countermeasure, higher eukaryotes have developed strategies for eliminating mitosis-incompetent cells, one of which is mitotic catastrophe. Mitotic catastrophe is driven by a complex and poorly understood signalling cascade but, from a functional perspective, it can be defined as an oncosuppressive mechanism that precedes (and is distinct from) apoptosis, necrosis or senescence. Accordingly, the disruption of mitotic catastrophe precipitates tumorigenesis and cancer progression, and its induction constitutes a therapeutic endpoint.
_____
Genome doubling perturbs DNA packing and promotes cancer development, a 2023 study:
Cells in which the whole genome has been doubled do not upscale protein synthesis to cope with the increase in DNA. Instead, a shortage of proteins that regulate the packing of DNA in the nucleus leads to poor segregation of DNA structures, which eventually contributes to the development of cancer.
______
______
Section-17
Cancer pathophysiology:
Regardless of difference in types of cancer histologically and physiologically, there is existence of a common pathophysiological process of malignant tumors or cancer development in the organism. The commonly accepted basis of the pathogenesis of cancer is the damage to the genetic apparatus of cells (such as mutation, disturbance of gene expression, activation of tumor promoter gene, inactivation of tumor suppressor genes, etc.). At the cellular level, the development of cancer is viewed as a multi-step process involving mutation and selection for cells with progressively increasing capacity for proliferation, survival, invasion, and metastasis.
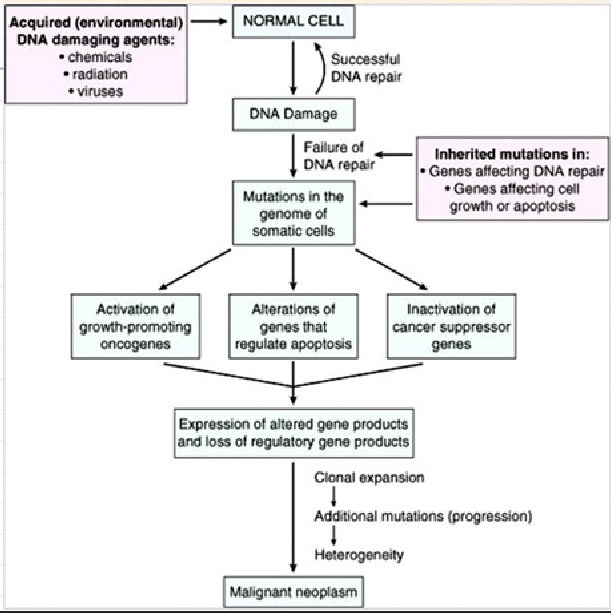
Figure above shows pathogenesis of cancer.
First step: Mutation and tumor initiation
- Genetic alteration leads to mutation in a single cell which results into abnormal proliferation of that cell known as tumor cell.
Second step: Cell proliferation and Tumor progression
- Tumor progression continues as additional mutations occur within cells of the tumor population.
- The mutated cells have some selective advantage over normal cell as such cells shows rapid growth and division. The descendants of a cell bearing such additional mutation will consequently become dominant within the tumor population
Third step: Clonal selection and malignancy
- Cell proliferation of tumor then leads to new clone of tumor cells with increased growth rate or other properties (such as survival, invasion, or metastasis) that confer a selective advantage. The process is called clonal selection.
- Clonal selection continues throughout tumor development, so tumors continuously become more rapid-growing and increasingly malignant.
For example: In colon cancer, the earliest stage in tumor development is increased proliferation of colon epithelial cells. A clonal selection occurs in which, a single cell within these proliferative cell population give rise to a small benign neoplasm. Further rounds of clonal selection lead to the growth of benign neoplasm with increase in size and proliferative potential resulting in malignant carcinoma. The cancer cells then continue to proliferate and spread through the connective tissues of the colon wall. Eventually the cancer cells penetrate the wall of the colon and invade other abdominal organs, such as the bladder or small intestine. In addition, the cancer cells invade blood and lymphatic vessels, allowing them to metastasize throughout the body.
Fourth step: Metastasis
- Metastasis is a complex process in which cancer cells break away from the primary tumor and circulate through the bloodstream or lymphatic system to other sites in the body.
- At new sites, the cells continue to multiply and eventually form additional tumors comprised of cells that reflect the tissue of origin.
- The ability of tumors, such as pancreatic cancer and uveal (iris, ciliary body, or choroid of eye) cancers, to metastasize contributes greatly to their lethality.
Many fundamental questions remain about the clonal structures of metastatic tumors, phylogenetic relationships among metastases, the scale of ongoing parallel evolution in metastatic and primary sites, how the tumor disseminates, and the role that the tumor micro-environment plays in the determination of the metastatic site.
_____
_____
Growth factors:
Although structurally altered genes, classified as oncogenes or tumor suppressor genes, are key mediators of neoplasia, the role of unaltered genes is not to be dismissed and is likely equally important in carcinogenesis. Signaling proteins of all kinds may drive the oncogenic process through abnormal signaling: abnormal in time, duration, or intensity; abnormal tissue expression; or abnormal subcellular compartment localization. The regulation of growth in complex organisms requires specialized proteins for the normal growth, maturation, development, and function of cells and specialized tissue. The complexity of the human organism requires that these proteins be expressed at precisely coordinated points in space and time. An essential component of this regulation is the system of hormones, growth factors, and growth inhibitors. On binding to specific receptor proteins on the cell surface or in the cytoplasm, these factors lead to a complex set of signals that can result in a variety of cellular effects, including mitogenesis, growth inhibition, changes in cell cycle regulation, apoptosis, differentiation, and induction of a secondary set of genes. The actual end effects are dependent not only on the particular type of interacting factor and receptor but also on the cell type and milieu in which factor–receptor coupling occurs. This system allows for cell-to-cell interactions, whereby a factor secreted by one cell or tissue can enter the bloodstream and influence another set of distant cells (endocrine action) or act on adjacent cells (paracrine action). An autocrine action is also possible when a cell produces a factor that binds to a receptor on or in the same cell. Altered concentration of these growth factors as well as overexpression or mutations of the receptors can change the signaling behavior, contributing to a malignant phenotype. Only a subset of growth factor receptors are proto-oncogenes. However, many additional growth factors and growth factor receptors appear to be important in tumor growth and progression, although not classified as proto-oncogenes, because they serve tumorigenic causes without incurring mutations or without overexpression.
_
Growth factors, which generally considered as a subset of cytokines, refer to the diffusible signaling proteins that stimulate cell growth, differentiation, survival, inflammation, and tissue repair. They can be secreted by neighboring cells, distant tissues and glands, or even tumor cells themselves. Normal cells show a requirement for several growth factors to maintain proliferation and viability. Growth advantage is often found for the cells which secrete a growth factor.
Growth factor can exert their stimulation though endocrine, paracrine or autocrine mechanisms. Due to their short half-lives and slow diffusion in intercellular spaces, growth factors usually act locally. Typically, the signal transduction of growth factors is initiated by binding to their receptors on the surface of target cells. The specific instruction conveyed by a growth factor to a particular subpopulation of cells depends on the type of receptors, number of target cell, and the intracellular signal transduction subsequent to factor binding. Moreover, external factors such as the binding ability of a growth factor to extracellular matrices (ECM), ECM degradation, and concentration of the growth factor may have an effect on the ultimate response of a target cell to a specific growth factor.
_
There are a number of different growth factors. These include:
- epidermal growth factor (EGF) – controls cell growth
- vascular endothelial growth factor (VEGF) – controls blood vessel development
- platelet derived endothelial growth factor (PDGF) – controls blood vessel development and cell growth
- fibroblast growth factor (FGF) – controls cell growth
Each growth factor works by attaching to the corresponding receptor on the cell surface. For example, EGF binds to epidermal growth factor receptor (EGFR). Tyrosine Kinases are chemical messengers (enzymes) used by cells to control how they grow and divide. They act like an ‘on-off’ switch. When the growth factor attaches to the outside of the cell it switches the tyrosine kinase ‘on’. This signals the cell to divide as seen in the figure below:
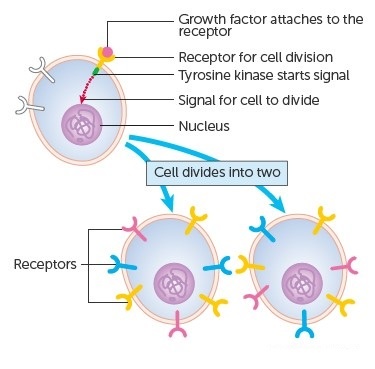
_
An important class of growth factor signaling molecules are the growth factor receptor tyrosine kinases (RTKs). A number of tyrosine kinase receptor families exist, and in experimental models most are capable of transforming cells if activated or overexpressed. Although all of these abnormalities are not necessarily seen in naturally occurring human tumors, these experimental data highlight the potential inherent in these proteins and the important role they may be playing in tumor cells despite lacking the oncogene label. Members of the HER family of RTKs are commonly mutated or amplified in human tumors and exemplify the important role of RTKs in human neoplasia. In many other tumors, they likely play an important role despite having a normal sequence and expression level. For example, HER1 (also called EGFR) is not mutated or overexpressed in colon cancers, but it is sometimes activated by autocrine signaling in the cancer cells, and EGFR-targeted therapies are used to treat this type of cancer. The platelet-derived growth factor (PDGF) receptors, fibroblast growth factor receptors, vascular endothelial growth factor receptors, and insulin-like growth factor receptor are all families of RTKs that function similar to HER family RTKs. These receptors are, in general, not reported to be mutated or amplified in human tumors. However, there is increased expression in many tumors or aberrant expression in tumors from tissue types that ordinarily would not be expected to express that receptor. Alternatively, excessive production of receptor ligands is due to a variety of mechanisms (i.e., loss of epigenetic silencing of the gene coding for the ligand or excessive gene transcription of the same gene). In experimental systems, each of these RTK systems has oncogenic potential, building a circumstantial case that they may be important players in human tumors.
_
Cellular signaling pathways control the proliferation, differentiation, survival, and migration of normal cells. But, dysregulation of these cells can cause tumors to form and grow. The overexpression or mutation of cell surface receptors and intracellular enzymes, along with the altered expression of growth factors, cytokines, and steroid hormones can promote the development and growth of malignancies, and trigger parenchymal cells to aid the development of tumors. Moreover, mutations of components within intracellular signaling pathways can cause activated pathways that are unresponsive to external signals working to inhibit proliferation and apoptosis.
_
Under normal physiological conditions, growth factor availability normalizes the balance between cell creation and division and cell death. This role in cellular homeostasis is often destabilized in cancer cases. Tumor cells can develop the ability to produce and secrete growth factors to stay alive, as well as make use of non-malignant cells to aid growth and avoid being identified by the immune system.
Growth factors bind transmembrane receptors, which contain inherent kinase activity. When activated, growth factors stimulate intracellular signaling pathways, including phosphoinositide 3-kinases (PI 3-K) and mitogen-activated protein kinase (MAPK) pathways as seen in the figure below.
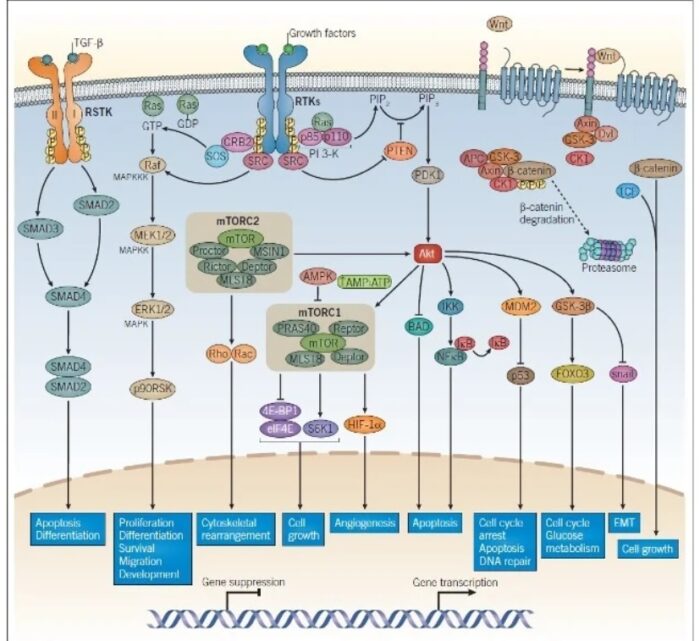
Figure above shows intracellular signaling pathways in Tumorigenesis and Cancer Progression.
In several human cancers, receptor tyrosine kinases (RTKs) are often affected by mutations causing upregulation of their signaling output. For example, the amplification or overexpression of the HER2/Neu/ERBB2 gene is recurrently seen in breast cancer cases. HER2 is part of the ErbB family of RTKs, made up of four members: epidermal growth factor receptor (EGFR or ErbB1), HER2 (ErbB2), ErbB3 and ErbB4. Intracellular signaling from ErbB homo- and heterodimers occurs through the PI 3-K and MAPK signaling pathways.
Type I insulin-like growth factor receptor (IGF1R) is significant in the development and endurance of several cell types and is universally expressed throughout the body. Dysregulation of IGF1R expression and signaling has been proved to drive tumor formation and progression. For example, studies have revealed that overexpression of IGF1R promotes the growth of tumors in mouse models and inhibits apoptosis in prostate cancer cells.
RTK growth factor receptors, TGF-β receptors, and Wnt signaling all play a role in tumorigenesis and cancer progression. Dimerization of growth factor receptors and TGF-β receptors occur upon ligand binding, enabling activation of the intracellular kinases on each receptor, leading to autophosphorylation. The phosphorylated residues on the cytoplasmic domain of the RTK bind adaptor proteins to initiate PI 3-K signaling and MAPK signaling. TGF-β binds the TGF-β receptor I/II dimeric complex and causes the activation of SMAD2 and SMAD3, which results in the association of SMAD2/SMAD4 and upregulation of TGF-β-responsive gene transcription. β-catenin undergoes proteasomal degradation in the absence of Wnt; upon Wnt binding, β-catenin is released from its complex and combines with TCF to promote Wnt-responsive gene transcription. These pathways regulate a wide range of cellular processes and encompass many of the major targets studied in cancer research.
Anaplastic lymphoma kinase (ALK) is an RTK first identified in anaplastic large cell lymphoma (ALCL) as part of the fusion protein NPM-ALK. It has been shown that downstream effectors of ALK tyrosine kinase activity include the Ras-ERK, PI 3-K-Akt, JAK-STAT, and NF-κB signaling pathways. In the absence of ligand binding, several ALK is inactive, and its expression promotes apoptosis. In contrast, when ALK is activated through either ligand binding or as part of an ALK fusion protein, rates of cell death is decreased. ALK promotes the development of tumors through overexpression and mutations leading to the development of certain functions. ALK is overexpressed in lung cancer, melanoma, and certain types of breast cancer, among others, while point mutations in the ALK kinase domain have been linked to the development of neuroblastoma.
Fibroblast growth factor (FGF) signaling is mediated by FGF receptors (FGFR). This signaling pathway is regulated by FGFR expression levels and the affinity of FGF for the different FGFR isoforms. In normal cells, FGF is a regulator in tissue homeostasis, tissue repair, and angiogenesis. Dysregulation of this pathway has been implicated in the development of tumors.
TGF-β is a growth factor that plays a key role in multiple pathways involved with cell adhesion, differentiation, motility, and death. In normal cells, TGF-β binds the serine/threonine kinase TGF-β receptors and makes cells less able to move through the cell cycle, as along with promoting cell death. Disruption of the TGF-β/SMAD pathway has been identified in a huge number of human cancers.
______
______
Nuclear receptors and cancer:
Nuclear receptors are receptors located inside the cell. These receptors are found either in the cytoplasm (Type I) or the nucleus (Type II) of a cell. Examples include: estrogen, glucocorticoids, thyroid hormone T3 or vitamins D and A. Nuclear receptors bind directly to DNA regulating the expression of adjacent genes; hence these receptors are classified as transcription factors. The regulation of gene expression by nuclear receptors often occurs in the presence of a ligand—a molecule that affects the receptor’s behavior. Ligand binding to a nuclear receptor result in a conformational change activating the receptor. The result is up- or down-regulation of gene expression.
_
Nuclear receptors (NRs) are transcription factors that orchestrate complex regulatory networks in many biological processes, including cell proliferation, metabolism, immunity, and development. The human NR superfamily, which consists of 48 members, includes receptors for steroid hormones, thyroid hormones, retinoic acid, vitamin D, fatty acids, and cholesterol metabolites (oxysterols). Members of the superfamily are encoded by genes that usually consist of eight exons and have a modular structure with four main functional domains, reflected in the exon/intron organization of the gene. Each NR achieves its transcriptional effects in concert with a number of coregulator proteins: coactivators, which are recruited to the ligand-bound receptor, and corepressors, which may be recruited when without the ligand or after being triggered by antagonists. The modes by which coregulators affect NR action are diverse and include promoting the recruitment of transcriptional machinery, remodeling chromatin, modifying histones, or acting as chaperones.
_
NR signaling pathways in cancer biology:
![]()
Nuclear receptors are critical for a variety of processes of both physiology and pathology, serving as sensors of stimuli and fine-tuning downstream molecular events that govern complex gene regulatory networks. The dysfunction of different nuclear receptors has been reported in different cancer types, rendering this family of proteins potential therapeutic targets for cancer patients. In particular, substantial advances have been made in the treatment and prevention of estrogen receptor (ER)+ breast cancer and the treatment of castration-resistant prostate cancer (CRPC) with the approval of small-molecule drugs that inhibit signaling by these receptors. Other NRs such as the glucocorticoid (GC) receptor (GR), the progesterone receptor (PR), retinoic acid receptors (RARs), and retinoid X receptors (RXRs) have also been extensively pursued as drug targets in cancer, in some cases resulting in further marketed drugs.
_
The actions of estrogen are fundamentally important in the development of breast cancer. In women, oophorectomy early in life offers substantial protection against the development of breast cancer, and in animal models mammary carcinogenesis is significantly retarded in the absence of estrogen. Approximately half of all breast cancers are dependent on estrogen for proliferation. Although these data clearly implicate the estrogen signaling pathway in breast carcinogenesis, specific abnormalities of the estrogen receptor (ER) are not seen in breast cancers; therefore, the ER does not qualify as a tumor suppressor protein or an oncoprotein. It is possible that, although the loss of certain tumor suppressor genes or activation of certain oncogenes leads to the development of breast cancer, continued ER function is essential throughout this process and without ER function it cannot proceed. Alternatively, it is possible that abnormal ER signaling, perhaps as a result of altered cofactors, cross-talk, or phosphorylation status, drives breast carcinogenesis. Although the mechanism by which estrogen and its receptor drive breast cancers has not yet been determined, its fundamental role in this disease is well established. Furthermore, treatments that work through inhibiting the production of the active ligand or that inhibit the function of the ER are the most effective therapies for breast cancer yet developed and are highly active in the prevention and treatment of breast cancer. The androgen receptor (AR), similarly, plays a critical role in the development of prostate cancer, although occasional activating mutations and amplification of the AR have been reported in prostate cancers. On the contrary, the ability of retinoids (ligands for retinoic acid receptors) that are well known to participate in the differentiation of a variety of tissues during development to cause the differentiation of certain tumors in tissue culture models has been exploited as a treatment approach for acute promyelocytic leukemia (APL). APL is characterized by a t(15;17) chromosomal translocation resulting in the fusion of the PML gene with the retinoic acid receptor-α (RAR-α) gene. The resulting fusion protein blocks the differentiation of hematopoietic progenitor cells and eventually leads to the development of APL. This fusion protein is not by itself transforming in experimental models and cannot be categorized as a classic oncogene or tumor suppressor gene, but it is etiologically involved in the pathogenesis of APL. Because the fusion protein contains the ligand-binding domain of RAR-α, it remains sensitive to ligand and treatment of patients with the ligand all-trans retinoic acid results in differentiation of tumor cells and complete remission in most patients with this disease.
______
______
Metastasis:
The metastatic process accounts for the vast majority of deaths from solid tumors, and therefore, an understanding of this process is critical for improvements in survival from cancer. The biology of metastasis is complex and requires multiple steps. The initial step involves cell migration and invasion through the extracellular matrix (ECM). The three major features of tissue invasion are cell adhesion to the basement membrane, local proteolysis of the membrane, and movement of the cell through the rent in the membrane and the ECM. Cells that lose contact with the ECM normally undergo programmed cell death (apoptosis induced by the loss of contact), and this process has to be suppressed in cells that metastasize. Another process important for many, but not necessarily all, metastasizing epithelial cancer cells is epithelial-mesenchymal transition (EMT). This is a process by which cells lose their epithelial properties and gain mesenchymal properties. This normally occurs during the developmental process in embryos, allowing cells to migrate to their appropriate destinations in the embryo. It also occurs in wound healing, tissue regeneration, and fibrotic reactions, but in all of these processes, cells stop proliferating when the process is complete. Malignant cells that metastasize often undergo EMT as an important step in that process but retain the capacity for unregulated proliferation. However, there is evidence that not all metastasizing cancer cells require EMT, and the exact role of EMT in different metastasizing cancer cells continues to be elucidated. Malignant cells that gain access to the circulation must then repeat those steps at a remote site, find a hospitable niche in a foreign tissue, avoid detection and elimination by host defenses including the immune system, and induce the growth of new blood vessels. Some metastatic cells occur as oligoclonal clusters, which appear to be more potent in establishing metastasis than single cells, perhaps, in part, through differential and cooperative effects in evading host defenses. The rate-limiting step for metastasis is the ability for tumor cells to survive and expand in the novel microenvironment of the metastatic site, and multiple host-tumor interactions determine the ultimate outcome as seen in the figure below.
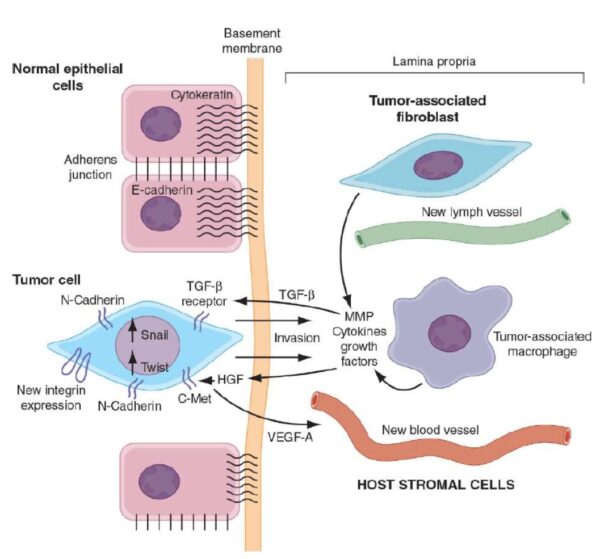
Figure above shows that oncogene signaling pathways are activated during tumor progression and promote metastatic potential. This figure shows a cancer cell that has undergone epithelial to mesenchymal transition (EMT) under the influence of several environmental signals. Critical components include activated transforming growth factor beta (TGF-β) and the hepatocyte growth factor (HGF)/c-Met pathways, as well as changes in the expression of adhesion molecules that mediate cell-cell and cell–extracellular matrix interactions. Important changes in gene expression are mediated by the Snail and Twist family of transcriptional repressors (whose expression is induced by the oncogenic pathways), leading to reduced expression of E-cadherin, a key component of adherens junctions between epithelial cells. This, in conjunction with upregulation of N-cadherin, a change in the pattern of expression of integrins (which mediate cell–extracellular matrix associations that are important for cell motility), and a switch in intermediate filament expression from cytokeratin to vimentin, results in the phenotypic change from adherent highly organized epithelial cells to motile and invasive cells with a fibroblast or mesenchymal morphology. EMT is thought to be an important step leading to metastasis in some human cancers. Host stromal cells, including tumor-associated fibroblasts and macrophages, play an important role in modulating tumor cell behavior through secretion of growth factors and proangiogenic cytokines, and matrix metalloproteinases that degrade the basement membrane. VEGF-A, -C, and -D are produced by tumor cells and stromal cells in response to hypoxemia or oncogenic signals and induce production of new blood vessels and lymphatic channels through which tumor cells metastasize to lymph nodes or tissues.
_
Important roles for LNs in anti-tumor immune responses:
Cancer treatment routinely involves taking out lymph nodes near the tumor in case they contain metastatic cancer cells. But new findings from a clinical trial by researchers at UC San Francisco and Gladstone Institutes shows that immunotherapy can activate tumor-fighting T cells in nearby lymph nodes. The study, published March 16, 2023 in Cell, suggests that leaving lymph nodes intact until after immunotherapy could boost efficacy against solid tumors, as only a small fraction of which currently respond to these newer types of treatments. Most immunotherapies are aimed only at reinvigorating T cells in the tumor, where they often become exhausted battling the tumor’s cancer cells. But the new research shows that allowing the treatment to activate the immune response of the lymph nodes as well can play an important role in driving positive response to immunotherapy. This work really changes our thinking about the importance of keeping lymph nodes in the body during treatment. Lymph nodes are often removed because they are typically the first-place where metastatic cancer cells appear, and without surgery, it can be difficult to determine whether the nodes contain metastases. Researchers have largely been working under the assumption that cancer immunotherapy works by stimulating the immune cells within the tumor but immunotherapy drugs are actually activating the lymph nodes. Rather than the immunotherapy pumping up the T cells in the tumor, T cells in the lymph nodes are likely the source for T cells circulating in the blood. Such circulating cells can then go into the tumor and kill off the cancer cells.
______
______
Tumour heterogeneity:
Tumour heterogeneity describes the observation that different tumour cells can show distinct morphological and phenotypic profiles, including cellular morphology, gene expression, metabolism, motility, proliferation, and metastatic potential. This phenomenon occurs both between tumours (inter-tumour heterogeneity) and within tumours (intra-tumour heterogeneity). A minimal level of intra-tumour heterogeneity is a simple consequence of the imperfection of DNA replication: whenever a cell (normal or cancerous) divides, a few mutations are acquired—leading to a diverse population of cancer cells. The heterogeneity of cancer cells introduces significant challenges in designing effective treatment strategies. Tumour heterogeneity has been observed in leukemias, breast, prostate, colon, brain, esophagus, head and neck, bladder and gynecological carcinomas, liposarcoma, and multiple myeloma.
_
The mutant cells that compose a single tumor are not genetically identical. Rather, cells obtained from different sites on a tumor will harbor common mutations as well as mutations that are unique to each sample. Genetic heterogeneity results from the ongoing acquisition of mutations during tumor growth. Each time a genome is replicated, there is a small but quantifiable probability that a mutation will spontaneously arise as a result of a replication error and be passed on to the cellular progeny. This is true in normal cells or in tumor cells. Any randomly chosen cell from the skin of one individual will harbor hundreds of genetic alterations that distinguish it from a different randomly chosen skin cell, and the same is true for all organs of self-renewing tissues. Tumors are actually less genetically heterogeneous than normal tissues; any two randomly chosen cells from a tumor of an individual will have fewer differences than any two randomly chosen cells from that individual’s normal tissues. The reason for this decrease in heterogeneity is clonal expansion, the fundamental feature of tumorigenesis. Every time a clonal expansion occurs, a genetic bottleneck wipes out heterogeneity among the cells that did not expand; these unexpanded cells either die or form only a minute proportion of the total cells in the expanding tumor. The mutations that vary between cells of a given tumor are invariably passenger mutations that arose since the last evolutionary bottleneck, that is, those mutations that arose during the expansion of the founder cell that gave rise to the final clonal expansion. In contrast, the passenger mutations that were present in the founder cell will be uniformly present in every cell in the tumor. In that respect, these passenger mutations are not heterogeneously distributed and are in fact uniformly present in virtually all cancer cells. These “clonal” mutations, i.e., present in all cells of the cancers, are the main source of Mutation-associated neoantigens MANAs that can be exploited through immune checkpoint inhibitors.
______
______
Cancer metabolism:
One of the distinguishing characteristics of cancer cells is that they have altered metabolism as compared with normal cells in supporting survival, their high rates of proliferation, and ability to metastasize. Complicating studies evaluating metabolic differences between normal and malignant cells is that there is heterogeneity in metabolism between different cells within a cancer. Malignant cells must focus a significant fraction of their energy resources into synthesis of proteins and other molecules (building blocks required for the production of new cells) while still maintaining sufficient ATP production to survive and grow. Although normal proliferating cells also have similar needs, there are differences in how cancer cells metabolize glucose and a number of other compounds including the amino acid glutamine as compared to normal cells in part because of genetic and epigenetic changes within cancer cells but also likely due to differences in the environments of cancer and normal cells.
_
Normal cells typically generate only about 30% of energy from glycolysis, whereas most cancers rely on glycolysis for energy production (Warburg effect). But a minority of cancer types rely on oxidative phosphorylation as the primary energy source, including lymphoma, leukemia, and endometrial cancer. Even in these cases, however, the use of glycolysis as an energy source rarely exceeds 60%. A few cancers use glutamine as the major energy source, partly because it provides nitrogen required for nucleotide (DNA, RNA) synthesis. Cancer stem cells often use oxidative phosphorylation or glutamine as a primary energy source.
_
Many cancer cells utilize aerobic glycolysis (the Warburg effect) (Figure below) to metabolize glucose, leading to increased lactic acid production, whereas normal cells utilize oxidative phosphorylation in mitochondria under aerobic conditions, a much more efficient process for generating ATP for energy utilization but one that does not produce the same level of building blocks needed for new cells.
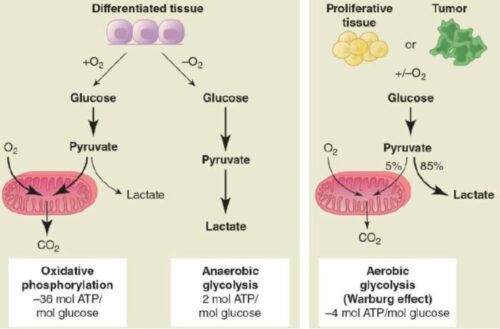
Figure above shows Warburg versus oxidative phosphorylation.
In most normal tissues, the vast majority of cells are differentiated and dedicated to a particular function within the organ in which they reside. The metabolic needs are mainly for energy and not for building blocks for new cells. In these tissues, ATP is generated by oxidative phosphorylation that efficiently generates about 36 molecules of ATP for each molecule of glucose metabolized. By contrast, proliferative tumor tissues, especially in the setting of hypoxia, a typical condition within tumors, use aerobic glycolysis to generate energy for cell survival and generation of building blocks for new cells.
_
One consequence is increased glucose uptake and utilization by cancer cells, a fact utilized in fluorodeoxyglucose (FDG)-positron emission tomography (PET) scanning to detect tumors. A number of proteins in cancer cells, including cMYC, HIF1, RAS, p53, pRB, and AKT, are involved in modulating glycolytic processes and controlling the Warburg effect. Although these pathways remain difficult to target therapeutically, both the PI3K pathway with signaling through mTOR and the AMP-activated kinase (AMPK) pathway that inhibits mTORC1 (a protein complex that includes mTOR) are important in controlling the glycolytic process and thus provide potential targets for inhibiting this process. An inhibitor of mTOR is approved for use against RCC (temsirolimus), and another inhibitor (everolimus) has activity against breast and neuroendocrine cancer and RCC. Other mTOR inhibitors are in trials, and modulators of AMPK are being investigated. The inefficient utilization of glucose by malignant cells also leads to a need for alternative metabolic pathways for other compounds as well, one of which is glutamine. Similar to glucose, this provides both a source for structural molecules as well as energy production. Similarly to glucose, glutamine is also inefficiently utilized by cancer cells. Cancer cells can also take up nutrients released by surrounding cells and tissues, increasing the complexity of successfully therapeutically inhibiting metabolism in cancer.
_
Mutations in genes involved in the metabolic process occur in a number of cancers. Among the most frequently found to date are mutations in isocitrate dehydrogenases 1 and 2 (IDH1 and IDH2). These have been most commonly seen in gliomas, AMLs, and intrahepatic cholangiocarcinomas. These mutations lead to the production of an oncometabolite (2-hydroxyglutarate [2HG]) instead of the normal product α-ketoglutarate. Although the exact mechanisms of oncogenesis by 2HG are still being elucidated, α- ketoglutarate is a key cofactor for a number of dioxygenases involved in controlling DNA methylation. 2HG can act as a competitive inhibitor for α-ketoglutarate, leading to alterations in methylation status (primarily hypermethylation) of genes (leading to epigenetic changes) that can have profound effects on a number of cellular processes including differentiation. Inhibitors of mutant IDH1 and IDH2 are approved for treating IDH mutant AML and are in clinical trials for glioblastomas and cholangiocarcinomas. Much needs to be learned about the specific differences in metabolism between cancer cells and normal cells; however, even with the currently limited state of knowledge, modulators of metabolism are being tested clinically. The first of these is the antidiabetic agent metformin, both alone and in combination with chemotherapeutic agents. Metformin inhibits gluconeogenesis and may have direct effects on tumor cells by activating AMPK, a serine/threonine protein kinase that is downstream of the LKB1 tumor suppressor, and thus inhibiting mTOR complex 1 (mTORC1). This leads to decreased protein synthesis and proliferation. Studies to date have not yet established metformin to have a clear role as an anticancer agent.
_
Cancer cells are notorious for their ability to divide uncontrollably and generate hordes of new tumor cells. Most of the fuel consumed by these rapidly proliferating cells is glucose, a type of sugar. Scientists had believed that most of the cell mass that makes up new cells, including cancer cells, comes from that glucose. However, MIT biologists have now found, to their surprise, that the largest source for new cell material is amino acids, which cells consume in much smaller quantities. The findings offer a new way to look at cancer cell metabolism, a field of research that scientists hope will yield new drugs that cut off cancer cells’ ability to grow and divide. To determine where cells, including those in tumors, were getting the building blocks they needed, the researchers grew several different types of cancer cells and normal cells in culture dishes. They fed the cells different nutrients labeled with variant forms of carbon and nitrogen, allowing them to track where the original molecules ended up. They also weighed the cells before and after they divided, enabling them to calculate the percentage of cell mass contributed by each of the available nutrients.
Although cells consume glucose and the amino acid glutamine at very high rates, the researchers found that those two molecules contribute little to the mass of new cells — glucose accounts for 10 to 15 percent of the carbon found in the cells, while glutamine contributes about 10 percent of the carbon. Instead, the largest contributors to cell mass were amino acids, which make up proteins. As a group, amino acids (excluding glutamine) contribute the majority of the carbon atoms found in new cells and 20 to 40 percent of the total mass.
It remains something of a mystery why proliferating human cells consume so much glucose. Consistent with previous studies, the researchers found that most of the glucose burned by these cells is excreted as lactate. This led researchers to conclude that the importance of high glucose consumption is not necessarily the manipulation of carbon that allows you to make cell mass.
_______
_______
Section-18
Cancer stem cell (CSC):
_
The origin of cancer cells:
Cancer is described as a proliferative, invasive, and metastatic disease that is caused by an accumulation of genetic abnormalities that randomly produce a malignant cell. Such abnormalities can be induced by chemical carcinogens, chronic inflammation, exposure to radiation, or by genetic predisposition. Cancer originates when normal cells accumulate DNA mutations over time and lose the ability to grow and proliferate in a regulated manner, leading to unrestricted cell proliferation. Phenotypically and functionally, cancer cells are abnormal and unstable and show inter- and intra-tumor heterogeneity. Inter-tumor heterogeneity is manifested as the difference in tumor composition between different individuals for the same cancer type; while intra-tumor heterogeneity is described as differences between tumor cells inside of the same tumor. Cancer cells can arise in almost any tissue but they are most commonly found in the breast, ovary, prostate, liver, stomach, pancreas, lung, brain, and bone marrow. Because of differences in genetic composition and oncogenic signaling, tumors in different tissues exhibit different behaviors. Pancreatic tumors, for example, have a tendency to be highly aggressive, while prostate tumors are more confined and easier to treat.
_
Cancer Stem Cell (CSC):
The paradoxical nature of some cancers gave rise to the notion of a “cancer progenitor” or a CSC as far back as the 1960s. The best illustration for this concept was CML, where the bulk of the tumor consists of terminally differentiated neutrophils that are incapable of recreating the malignancy. However, it was apparent that there were multipotent stem cells in this tumor population. In 1994, Dick’s group proved conclusively that another type of leukemia, acute myelogenous leukemia (AML), was a hierarchically organized disease in which a small number of cells that were undetectable by conventional methods could be isolated from patients and made to recapitulate the full spectrum of the disease in an animal model. This gave rise to the CSC or “tumor-initiating cell” hypothesis, which is based on the concept that tumors are hierarchically organized into a subpopulation of cells that retain or acquire the capacity for self-renewal and are capable and responsible for initiating and maintaining the tumor. Another subpopulation of cells that consists of the CSC progeny undergo partial to complete differentiation and lose the capability to support the tumor, albeit they still contribute to the morbidity of cancer. This hypothesis fundamentally altered the way we understand cancer but also gave rise to a debate regarding how widely this model applies. The competing hypothesis is based on a model where all the cells in a tumor possess an equal capacity for self-renewal and is commonly referred to as the stochastic model. According to this model, the process of cancer is driven entirely (or almost entirely) by environmental selection of favorable mutations; this model would necessarily predict that cancer is an inevitable outcome for multicellular organisms, and few, if any, long-lived animals would reach reproductive age. Thus this model must, by necessity, invoke the existence of protective mechanisms that are independent of cancer risk (e.g., efficient DNA repair mechanisms and immune surveillance).
It is possible that the two models represent a continuum dependent on the extent to which CSCs undergo asymmetric versus symmetric divisions. Under conditions in which CSC divisions are primarily asymmetric, few CSCs would be apparent and the population would achieve a hierarchical organization, whereas under conditions in which CSCs underwent symmetric divisions, virtually every cell in the tumor would have CSC-like properties and the organization would be more consistent with a stochastic model. The prevailing opinion is that CSCs exist and are characterized both by peculiar phenotypes and defined sets of mutations of a small number of genes. Other mutations then endow their progeny with limited or extensive capacity to undergo programmed differentiation, thus resulting in the distinct clinical phenotypes that characterize acute and chronic leukemias or high-grade and low-grade solid tumors. The origin of CSCs is among the most important contemporary topics of investigation. CSCs may arise from mutations that occur in bona fide stem cells, they may arise by “de-differentiation” of somatic cells that acquire mutations that endow them with stem cell–like properties, or they may develop by fusion of a transformed cell and a bone marrow–derived stem cell. In companion animals, putative CSCs have been identified in hemangiosarcoma, osteosarcoma, brain tumors, and possibly lymphoma.
_
The cancer stem cell hypothesis proposes that the different kinds of cells in a heterogeneous tumor arise from a single cell, termed Cancer Stem Cell. Cancer stem cells may arise from transformation of adult stem cells or differentiated cells within a body. These cells persist as a subcomponent of the tumor and retain key stem cell properties. They give rise to a variety of cells, are capable of self-renewal and homeostatic control. Furthermore, the relapse of cancer and the emergence of metastasis are also attributed to these cells. The cancer stem cell hypothesis does not contradict earlier concepts of carcinogenesis. The cancer stem cell hypothesis has been a proposed mechanism that contributes to tumour heterogeneity.
_
On a cellular level, cancer progression can be divided in different stages: tumor initiation, tumor progression, angiogenesis, and metastasis. When these heterogeneous tumors are put under selection pressure by chemotherapy, a specific sub-population of resistant cells can selectively take over, allowing this sub-class to dominate the tumor. Recently, it was identified that a small part of that heterogeneous population is composed of stem-like progenitor cells or CSCs that could originate from normal or stem cell mutations initiated by changes in the environment, by chronic inflammation, or by epithelial-to-mesenchymal transformation (EMT) as seen in the figure below.
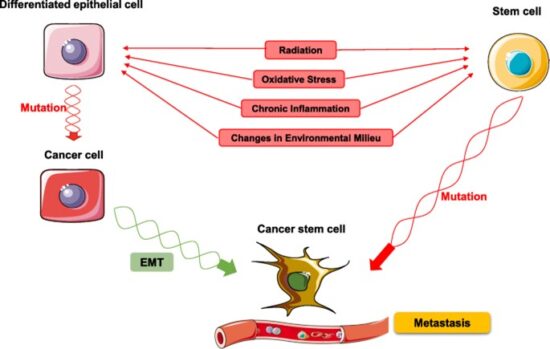
When epithelial cells are exposed to stressors such as radiation, oxidative stress, chronic inflammation, or changes in its environmental milieu, mutations in DNA can occur. While stem cells can mutate directly into CSCs, epithelial cells require a two-step process: first, the initial stress causes it to mutate into a cancer cell; then, it can undergo epithelial-mesenchymal transition (EMT) to become a cancer stem cell capable of metastasis. Mutations in stem cell can also occur by gradual accumulation of aging-induced genetic changes during lifetime.
_
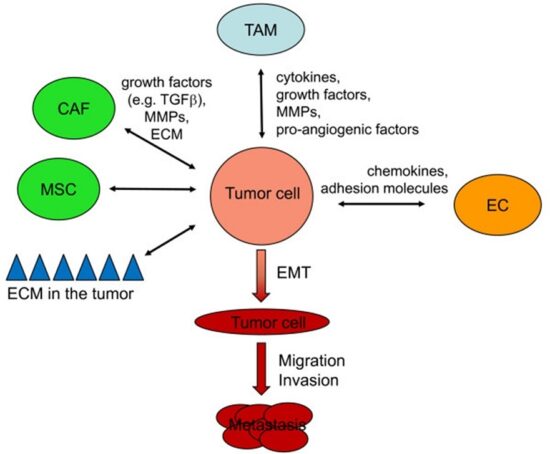
Figure above shows reciprocal interactions of tumor cells with the extracellular matrix (ECM), tumor-associated macrophages (TAM), carcinoma-associated fibroblasts (CAF), mesenchymal stem cells (MSC), and endothelial cells (EC). These interactions are mediated by direct cell-to-cell contact and/or the release of cytokines, chemokines, growth factors, matrix metalloproteases (MMPs), and ECM proteins. Eventually this results in epithelial-mesenchymal transition (EMT) of tumor cells, their migration, invasion, and dissemination to distant organs and the formation of metastases.
_
The lineages of cells in which DNA alterations accumulate are difficult to trace, but two recent lines of evidence suggest that normal stem cells may be the cells of origin in cancers.
First, there exists a highly positive correlation between the risk of developing cancer in a tissue and the number of normal stem cell divisions taking place in that same tissue. The correlation applied to 31 cancer types and extended across five orders of magnitude. This strongly suggests that the main factor in cancer initiation is the fact that “normal” stem cells divide, which implies that cancer originates in normal, healthy stem cells.
Second, statistics show that most human cancers are diagnosed in older people. A possible explanation is that cancers occur because cells accumulate damage through time. DNA is the only cellular component that can accumulate damage over the entire course of a life, and stem cells are the only cells that can transmit DNA from the zygote to cells late in life. Other cells, derived from stem cells, do not keep DNA from the beginning of life until a possible cancer occurs. This implies that most cancers arise from normal stem cells.
_
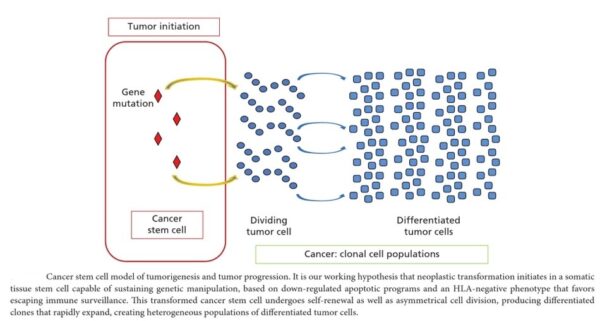
Cancer stem cells (CSCs) is a sub-population of cells that share many common characteristics with somatic stem cells (SSCs). The most critical properties of CSCs are their self-renewal ability and their capacity for differentiation into heterogeneous populations of cancer cells. Although CSCs only constitute a low percentage of the total tumor mass, these cells can regrow the tumor mass on their own. Initially identified in leukemia, CSCs have subsequently been found in cancers of the breast, the colon, the pancreas, and the brain. Common genetic and phenotypic features found in both SSCs and CSCs, including upregulated signaling pathways such as Notch, Wnt, Hedgehog, and TGF-β. These pathways play fundamental roles in the development as well as in the control of cell survival and cell fate and are relevant to therapeutic targeting of CSCs. The differences in the expression of membrane proteins and exosome-delivered microRNAs between SSCs and CSCs are also important to specifically target the stem cells of the cancer.
_
Different models are proposed to explain how established cancers propagate themselves. Classically, these include the CSC model, the clonal evolution model (also called the stochastic model) and the interconversion model. Each of these models explains anecdotal and experimental observations that many cancers. In the broad consideration of cancer propagation, the cancer stem cell model, the clonal evolution model and the interconversion model should not be thought of as mutually exclusive. Cancers that follow a CSC model contain rare tumorigenic cells that may undergo clonal evolution and/or may interconvert between different states of malignant behaviour and therapy resistance.
_
The strongest evidence for the CSC theory comes from studies in acute myelogenous leukemia (AML), when Bonnet and Dick performed serial transplantation experiments to show that only rare cells with high self-renewal capacity isolated from AML patients could initiate leukemia in murine models. Later the cell responsible for tumor initiation was identified by its phenotype as CD34+CD38−, a remarkably common phenotype of a normal hematopoietic stem cell (HSC). Additional studies have supported the idea of CSCs and currently this idea is becoming commonly accepted. While CSCs might represent a small fraction of the cells in the heterogeneous tumors, they likely play a fundamental role in cancer initiation, progression, and metastasis as well as in therapy failure. CSCs are also called tumor-initiating cells since they possess several important properties of SSCs: self-renewal, unlimited proliferation potential, slow replication, resistance to drugs, and the capacity to differentiate, giving rise to daughter cells, which make up the bulk of the tumor. While CSCs can initiate tumor formation, their daughter cells are incapable of forming new tumors by metastasis. There is some controversy regarding the origin of CSCs: while in most instances they may originate from normal stem cells, there are some CSCs that can arise from somatic cells. Since their origin is not clearly understood, the term tumor-initiating cells have been used to define these cells. In general, CSCs are considered the seed of the tumor mass which also promotes its growth.
Intriguingly, Scadden showed that somatic cells, which give rise to cancer cells, do not need to have stem cell properties but that a stem-like phenotype could be acquired during the process of tumorigenesis, which is further an indication of the tumor cells ability to adapt to environmental factors.
_
Consistent with the cancer stem cell model, certain cancers (including some germ cell cancers and some leukemias) have been recognized for decades to include neoplastic cells that differentiate into post-mitotic derivatives. Germ cell cancers give rise to highly differentiated cells, such as those that make neural tissue (Illmensee and Mintz, 1976). Some leukemias give rise to highly differentiated hematopoietic cells (Fearon et al., 1986, Barabe et al., 2007). These cancers are obviously hierarchically organized. The more controversial question raised by recent studies is whether many other cancers exhibit a similar hierarchical organization, even when overt differentiation is not evident among the cancer cells.
_
Compelling data support the cancer stem cell model in various human cancers including malignant germ cell cancers (Kleinsmith and Pierce, 1964, Illmensee and Mintz, 1976), leukemias (Lapidot et al., 1994, Bonnet and Dick, 1997), breast cancers (Al-Hajj et al., 2003), brain cancers (Singh et al., 2004), and colon cancers (Dalerba et al., 2007, O’Brien et al., 2007, Ricci-Vitiani et al., 2007). In each case, only small subpopulations of cells can transfer disease upon transplantation into immunocompromised NOD/SCID mice, and markers have been identified that distinguish the leukemogenic/tumorigenic cancer cells from the bulk populations of nonleukemogenic/ tumorigenic cells. The ability to predict which cells are tumorigenic based on marker expression indicates that the tumorigenic cells are intrinsically different from nontumorigenic cancer cells. Yet, no clear morphological distinction was found between tumorigenic and nontumorigenic breast cancer cells (Al-Hajj et al., 2003), implying that differentiation need not be overt for the cells to be hierarchically organized. The observation that tumorigenic cells have tended to be rare in the cancers found so far to follow a cancer stem cell model implies that epigenetic differences distinguish tumorigenic from nontumorigenic cells because it is implausible that only rare cancer cells have a genotype permissive for extensive proliferation. Tumorigenic cancer stem cells also form phenotypically diverse nontumorigenic cells, recapitulating at least some of the heterogeneity in the tumors from which they derive.
_
Despite strong data supporting the stem cell model in some cancers, it is important to acknowledge a number of caveats. There is no direct evidence in these cancers that tumorigenic cells differ from nontumorigenic cells as a result of epigenetic rather than genetic differences. Moreover, the conclusion that transplanted cancer stem cells can recapitulate the heterogeneity of the tumors from which they derive is based on limited analyses of only two or three surface markers. It has not yet been determined whether there is also genetic heterogeneity within the primary tumors that is not recapitulated after cancer stem cell transplantation. It therefore remains possible that the functional and phenotypic diversity within these cancers is underestimated and partially genetically determined.
_
The cancer stem cell model has been carefully tested in only a small subset of cancers. Although this model is often assumed to apply widely to other cancers, recent data demonstrate that leukemogenic/tumorigenic potential is a common attribute of cells in some cancers (Kelly et al., 2007, Williams et al., 2007, Quintana et al., 2008) making this a questionable assumption. Second, the NOD/SCID mouse transplantation assay, which has been the source of most of the compelling data supporting the cancer stem cell model, dramatically underestimates the frequency of human cancer cells with tumorigenic potential in some cancers (Quintana et al., 2008). This suggests the need to re-evaluate some of the evidence supporting the model using assays that are more permissive for the engraftment of human cancer cells. If some cancers that are currently thought to follow a cancer stem cell model actually have common tumorigenic cells and heterogeneity is generated through genetic and epigenetic mechanisms, the progression of these cancers may be more accurately described by the clonal evolution model (Nowell, 1976) (Table below).
_
Models to Explain Cancer Cell Heterogeneity:
|
Cancer Stem Cell Model |
(Stochastic) Clonal Evolution Model a |
|
|
Frequency of cancer cells with tumorigenic potential |
Rare to moderate |
High |
|
Phenotype of cancer cells |
Heterogeneous |
Heterogeneous or homogeneous |
|
Tumor organization |
Hierarchical |
Not necessarily hierarchical |
|
Intrinsic differences between tumorigenic and nontumorigenic cells |
Stable, epigenetic |
Unstable, epigenetic or genetic |
|
Rational approach to therapy |
Possible to target only tumorigenic cells |
Target most or all cells |
|
Compelling clinical evidence |
Germ lineage cancers |
High-grade B cell lymphoblastic leukemia b |
-a The clonal evolution model holds that genetic and epigenetic changes occur over time in individual cancer cells, and that if such changes confer a selective advantage they will allow individual clones of cancer cells to out-compete other clones. Clonal evolution can lead to genetic heterogeneity, conferring phenotypic and functional differences among the cancer cells within a single patient. Note that the clonal evolution and cancer stem cell models are not mutually exclusive in cancers that follow a stem cell model, as cancer stem cells would be expected to evolve by clonal evolution. However, heterogeneity in cancers that do not follow a cancer stem cell model (not hierarchically organized into epigenetically distinct tumorigenic and nontumorigenic populations) could be determined entirely by clonal evolution.
-b B cell lymphoblastic leukemias have extraordinarily high frequencies of leukemogenic cells that are not hierarchically organized in a mouse model (Williams et al., 2007) and appear homogeneous by histopathology in patients, yet heterogeneity can arise in sensitivity to therapy through clonal genetic changes.
______
______
Key properties of CSCs:
CSCs share the same cellular and molecular mechanisms that regulate SSCs; however, CSCs lack the necessary control system to prevent uncontrolled proliferation. While the specific origin of CSCs is still under debate, evidence suggests that they originate from stem cells that failed to control proliferation under abnormal circumstances. Other proposed origins suggest that CSCs could arise from cell-cell fusion between cancer cells and adult stem cells, gene transfer between somatic and cancer cells, or mutations in stem cells. In addition, transformation could occur during the process of tissue regeneration in response to inflammation, infection, toxin exposure, and/or metabolic processes which could cause mutations.
CSCs have been identified in several solid tumors based on the expression of certain CSC surface markers. Until now, CSCs have been identified by surface markers that are common between different cancer types: CD24, CD29, CD44 CD90, CD133, aldehyde dehydrogenase 1 (ALDH1), and epithelial-specific antigen (ESA). Depending on the type of tissue from which they originate, they can express a variety of markers for each type of CSCs. Most importantly, the expression of these markers can be used for specific therapeutic targeting of CSCs.
_
Evidence from a xenotransplantation experiment of human brain tumors into non-obese diabetic severe combined immunodeficiency (NOD/SCID) mouse brains did demonstrate the presence of CSCs in the tumor fraction responsible for tumor regeneration. Other studies also report that certain types of cancers, such as hepatocellular carcinoma (HCC), can be derived from SSCs. HSCs originate from self-renewing progenitors in the bone marrow where they mostly reside; however, they also can be found in peripheral blood. Clinical and genetic evidence suggest that certain types of leukemia are driven by genetic mutation in HSCs. These mutations give rise to leukemia stem cells, also called CSCs of the blood. Additionally, in a NOD/SCID murine model of leukemia, phenotypic differences between leukemia stem cells and HSCs were shown in separate studies. Recent efforts uncovered surface markers that are exclusive for AML stem cells such as CD123, TIM3, CD47, CD96, CLL-1, and IL-1 receptor accessory protein. On the other hand, AML stem cells rarely express CD90 and CD117 on their surface and several studies have reported that CD123 was predominantly expressed in a subset of AML stem cells characterized by CD34+/CD38− phenotype. These basic differences in the phenotype of CSCs could be essential to providing new avenues for the development of targeted cancer therapeutics.
_
CSCs are commonly confused with cancer-initiating cells because of their stem cell-like properties, where both are characterized by elevated expression of the stem cell surface marker CD133. When cancer-initiating cells receive the first cancer-causing mutation, they are hypothesized to be different from SSCs; however, they show several similarities such as low proliferative rates, high self-renewal, and resistance to chemotherapy and radiation. Currently, it is not clear whether CSCs originate from cancer-initiating cells or if both cell types have the same origin. However, both cells support tumor initiation and propagation.
_
-1. Environmental milieu and chronic inflammation support CSCs:
Cell microenvironment is fundamental for cell growth, fate, and interaction with other cells in response to a specific stimulus. Recent studies have confirmed that the microenvironment can support generation and growth of solid tumors, and it is possible that alterations in paracrine signals from niche cells could initiate or enhance tumor formation from SSCs. These signals function as a stimulus to induce activation, differentiation, proliferation, and/or cell death. Moreover, these environmental stimuli are a part of a greater structure called “stem cell niche”. This niche refers to a specific microenvironment inside a discrete anatomic location where SSCs are found in an undifferentiated and self-renewable state. These niches have been observed in different mammalian epithelial tissues, in the gastro intestinal tract, and in the neural and hematopoietic system where they regulate stem cell fate by directing cell-cell interaction and secretion factors. Soluble factors secreted from primary tumors can stimulate the recruitment of cells to the niche. Growth factors such as VEGF, TGF-β, and TNF-α have been identified as the major factors secreted from primary tumors which promote angiogenesis.
Soluble factors released in the microenvironment strongly influence the growth of a primary tumor, where its growth is further enhanced by changes in the niche environment. CSCs reside in these niches which are responsible for maintaining the self-renewal capacity and undifferentiated state of CSCs. These niches maintain the main properties of CSCs by preserving their phenotypic plasticity, protecting the cells against the immune system and low nutrient availability, resisting oxidative stress, detoxifying drugs via ATP-binding cassette transporters, and promoting metastasis. These niches also favor the recruitment of more cells to induce inflammation by secreting factors (cytokines and chemokines). The secreted factors facilitate the formation of secondary and tertiary tumors, where CSCs disseminate through the stroma into the bloodstream and induce metastasis.
Another important environmental factor driving tumor progression is chronic inflammation. This condition is hypothesized to be one of the principal factors in CSCs expansion and tumor dissemination. This inflammatory response can be initiated by the activation of toll-like receptors (TLRs), stimulated by pathogen-associated-molecular patterns (PAMPs) of carcinogenic microbes or by products released from cancer cells. Consequently, nuclear factor kappa B (NF-κB) is activated inducing an inflammatory response that could increase self-renewal activity in cancer cells. It was shown that in CD44+/MyD88+ epithelial ovarian CSCs, the TLR2/MyD88/NF-κB signaling can support its growth and self-renewal by the upregulation of stemness-associated genes. Another TLR that seems to promote the expression of stemness-associated genes is TRL3. For example, Jia et al. showed that poly(I:C) enhances stemness in cancer cells by the mutual activation of β-catenin and NF-κB signaling pathway. They demonstrated both in vitro and in vivo that breast CSCs, characterized by expression of CD44high/CD24−/low markers, possess stem cell-like properties and tumor-initiating capacity. They also established that TLR3 activation in breast cancer cells contributes to an enrichment of a subset of cells with the CSC phenotype.
Further evidence that supports the role of inflammation in cancer progression stems from observations that malignant tumors often develop at sites of chronic inflammation and tissue injury. Chronic viral hepatitis, general gastric inflammation, gastritis caused by Helicobacter pylori, inflammatory bowel disease, and several other chronic inflammation conditions are also shown to increase the risk of cancer development, and the induction of CSCs. Tumor stroma also contains activated fibroblasts, inflammatory cells, and nascent blood capillaries. The formation of such microenvironments facilitates induction of an inflammatory response that causes cell migration and epithelial cell proliferation. This results in tissue repair that can occasionally turn into uncontrolled cell proliferation and dissemination. O’Brien et al. showed that the ability of cancer cells to function as CSCs depends on how they respond to the self-renewal signals released in the environmental milieu. It was suggested that changes in the environmental milieu can lead to reprogramming of SSCs turning them into cancer stem cells after prolonged inflammation, infection, exposure to toxins, or autoimmune diseases. Some reports have shown that tumor environmental milieu provides the stimuli necessary for the transformation of SSCs by secreting TGF-β. This cytokine will enhance the transition from SSCs to CSCs by inducing zinc finger E-box-binding homeobox1 (ZEB1) transcription factor expression. ZEB1 contributes to cancer dissemination and the activation of epithelial-mesenchymal transition (EMT), a process that has been linked to cancer metastasis. Additional evidence suggests that ZEB1 is responsible for the maintenance of CSC-like phenotypes. Another example of inflammatory conditions affecting CSC development is hepatocellular progenitor cancer cells (HcPCs), which have been observed following chronic inflammation in the liver. HcPCs show a similar transcriptomic profile to the SSCs in the liver but these cells do not generate tumors. However, under chronic inflammation, interleukin-6 (IL-6) secretion stimulates HcPC growth in vivo and facilitates tumor progression. The role that chronic inflammation plays in the induction of different types of CSCs is still under investigation, but cytokines secreted by tumor-associated immune cells seem to activate the necessary pathways required by cancer cells to become cancer stem cell like.
_
-2. Immunoregulatory properties of CSCs:
It is well known that the immune system plays an important role in attacking tumor cells by detecting traits of malignant transformation by immune cells. Conversely, tumor cells can sometimes escape immune recognition, especially when progenitor cancer cells have already been established. It has been shown that CSCs are more tumorigenic than regular cancer cells found in the solid tumor and CSCs might evade the host immune response due to their phenotypic and functional properties, allowing them to endure and spread throughout the body. Tumor cells can evade the host immune response by different mechanisms, among them are the production of immunosuppressive factors, low expression of tumor antigens, lack of expression of human leukocyte antigens (HLAs), and co-stimulatory molecules. Immunosuppressive factors are produced in the tumor microenvironment where other immune cells also participate by releasing soluble factors that further immunosuppress the environment. The action of tumor-associated macrophages (TAM), an activated M2 polarized population, suppresses the inflammatory response and promotes tumor angiogenesis. It was shown that TAM co-localized with CD133+ glioma cells increased the invasive capability of these cells by secreting TGF-β1. Little is known about CSCs immune-regulatory properties but certainly they represent an important factor in the failure of cancer treatment. It was suggested that CSCs can escape natural killer (NK) cells recognition by entering into a latency stage and downregulating ULBP ligands that binds to and activates NK cells. Additionally, CSC can evade cytotoxic T cells by downregulating MHC-I and upregulating the expression of PD-L1, resulting in host immunosuppression.
A study compared the immunological properties of CSCs-like cells (CD44+) with tumor cells (CD44−) and demonstrated that CD44+ cancer stem-like cells have an immunosuppressive phenotype. This effect was probed by the inhibition of activated T cell proliferation, the induction of Treg polarization, and the suppression of Th1, which leads to the dysfunction of effector T cell and cytotoxic T cells. Another study showed that CSCs isolated from therapy-resistant tumors have pro-inflammatory tumorigenic properties; such CSCs can induce macrophage colony-stimulating factor production by activating interferon regulatory factor-5 (IRF-5) pathway. This study also found that they could induce several pro-inflammatory cytokines and chemokines such as TNF-α, IL-6, and IL-8, creating a tumorigenic microenvironment. Recent studies also suggest that, in addition to secreted cytokines, other secreted factors also contribute to downregulation of the immune response. For example, extracellular vesicle-mediated communication between tumor cells and immune cells may result in downregulation of the immune response to tumor cells. Bi et al. accentuated the role of IRF-5 in regulating tumor infiltration and found that the loss of IRF-5 expression in human ductal carcinoma correlates with disease stage and contributes to metastasis. Therefore, it appears that MSCs and CSCs share the same immune-regulatory properties. The difference is that MSCs use these properties in regeneration of damaged tissues and in immunomodulation, while CSCs use this property to endure in the tumor and evade the immune response. These characteristics also suggest that immune-suppression pathways could potentially be targeted for tumor clearance by immune response mediated mechanisms.
_
-3. Self-renewal activity and signaling pathways in CSCs:
Similar signaling pathways regulate the self-renewal activity in ESCs, SSCs, and CSCs. ESCs are pluripotent and can differentiate into any specialized cell in the body, while SSCs have more limited differentiation capacities. ESCs can be maintained in culture for long periods of time without losing their undifferentiated state. On the contrary, SSCs cultured in vitro differentiate rapidly, indicating that environmental and internal signals are fundamental for SSCs differentiation process and self-renewal. It is suggested that internal signals directed by genetic and epigenetic processes, and external signals composed of secreted cytokines and growth factors can change the fate of SSCs. The self-renewal activity of SSCs is highly regulated by different signaling pathways but this tight regulation is lost in CSCs. It has been shown that specific pathways such as Wnt/ β-catenin, Jack/Stat, TGF-β, Notch, and Sonic Hedgehog are deregulated in CSCs. Instead, CSCs have the ability to use these self-renewal pathways to drive tumor dissemination.
- Role of Wnt/β-catenin pathway in CSCs:
The Wnt signaling pathway belongs to a family of secreted glycoproteins that have several functions, including regulating proliferation, differentiation, and patterning throughout embryonic development. Other components of the Wnt pathway include molecules of the Wnt secretory machinery, Wnt co-receptors, new components of the β-catenin degradation machinery, and nuclear co-factors. Mutations in Wnt genes or defects in their signaling pathway cause specific developmental defects during embryonic development leading to certain human diseases and cancer during adulthood. Abnormal activation of the Wnt/β-catenin pathway is one of the most frequent abnormalities present during tumorigenesis. Abnormal expression of Wnt ligand proteins has been observed in different types of tumors in osteosarcoma, hematological malignancies, breast cancer, and non-small cell lung cancer. Malanchi et al. showed that β-catenin signaling is essential in maintaining CSC phenotype and its inhibition results in the loss of CSCs and total tumor regression indicating increased activation of β-catenin pathway in human squamous cell carcinoma. Moreover, Wnt signaling has been shown to be deregulated in leukemic stem cells compared to somatic hematopoietic stem cells. These results were obtained from a genome-wide expression analysis that compared the expression profile of highly enriched normal human HSCs and leukemic stem cells from patients with AML. In AML, deregulated activation of the Wnt/β-catenin pathway induces cell proliferation by turning on genes encoding oncoproteins and cell-cycle regulators. Furthermore, Wnt5a pathways seem to be involved in the regulation of CSCs, as reviewed by Zhou et al. which showed that there is a complex cross-talk between Wnt5a and specific receptors that are expressed in ESCs and that may be expressed or activated in CSCs but not in SSCs.
- STAT signaling deregulation is associated with CSCs induction:
Signal transducers and activators of transcription (STATs) involve tyrosine phosphorylation by Janus family tyrosine kinases (JAKs) that further allow STAT protein dimerization and nuclear translocation and, finally, expression of target genes. Gene encoding of the STAT family is localized on different chromosomes: in chromosome 2 (STAT1 and STAT4), in chromosome 12 (STAT2 and STAT6), and in chromosome 17 (STAT3 and STAT 5A/B). JAK-STAT signaling pathway regulates somatic cell differentiation, proliferation, immune response, and apoptosis. There is a role of JAK-STAT3 signaling pathway in promoting cancer through inflammation, obesity, stem cells, and pre-metastatic niche, and regulation of this pathway in tumors constitute an important target for therapeutics in the treatment of cancer. It is also found that STAT3 activation mediates the loss of androgen receptor expression, which promotes a stem-like cell phenotype in prostate cancer. STAT signaling pathway has also been shown to regulate both stem cell-renewal and tumorigenesis, and abnormal activation of the STAT pathway induces cell transformation and oncogenesis of many cancer types. This deregulation of STAT pathway can cause the inhibition of differentiation pathways and instead induces stem cell self-renewal. Moreover, it has been shown that STAT signaling plays a role in different stem cell niches in SSCs and CSCs. For example, STAT5 was shown to be activated in HSCs by the stem cell factors c-Kit and thrombopoietin, both fundamental factors for HSCs self-renewal. STAT5 has also been observed to be constantly expressed in different hematological and non-hematological malignancies. Hernandez-Vargas et al. suggested that a sub-population of sorted CD44+/CD24−, considered to be breast CSCs, constitutive activate JAK-STAT signaling pathway. Their data also support the concept that the expression of cancer stem-like pathways and the establishment and self-renewal properties of cancer stem cells are coordinated by epigenetic mechanisms.
- TGF-β pathway is involved in CSCs development and tumor progression:
The TGF-β pathway is another important pathway involved in embryonic development, in adult tissue homeostasis, and in the regulation of stemness of CSCs . TGF-β signaling is initiated upon TGF-β ligand interaction with type II and type I transmembrane serine/threonine kinase receptors on the cell surface which induces oligomerization of the receptor kinases and phosphorylation of the cytoplasmic signaling molecules Smad2 and Smad3 for the TGF-β/activin pathway. The activated Smad complexes are translocated into the nucleus together with other nuclear co-factors to regulate the transcription of target genes. Interestingly, the loss of function of certain Smads is observed during tumorigenesis of pancreatic and colorectal cancers as well as other cancer types. TGF-β is mostly known for its role as hepatic pro-fibrogenic cytokine predominantly produced by activated mesenchymal cells upon chronic liver damage, suggesting that it also participates in tissue repair and maintenance in SSCs. TGF-β induces Smad3-dependent nuclear accumulation of β-catenin in MSCs, and it was shown that TGF-β signaling regulates the differentiation fate of MSCs. Together with CSCs, TGF-β participates in the initiation and development of various tumors, and in the acquisition of CSC-like properties.
- Notch and Sonic hedgehog signaling are important in CSCs development:
Notch signaling pathway also plays an important role during embryogenesis and cancer development. Notch signaling is initiated by the interaction between a Notch ligand and a Notch receptor expressed on the surface of neighboring cells. In mammals, the Notch pathway is formed by five canonical type I transmembrane ligands, including Delta-like ligands (DLLs), DLL1, DLL3, and DLL4; two Jagged proteins, Jagged1 and Jagged2; and four Notch transmembrane receptors, Notch1–4. This interaction triggers a two-step proteolytic cleavage of the receptor, mediated by an ADAM/TACE (tumor necrosis factor alpha converting enzyme) metalloproteases. ADAM10 and ADAM17 interact with nuclear factors to regulate target gene expression that regulates cell differentiation. This pathway is mainly involved in cell to cell communication, tissue differentiation, and self-renewal of stem cells. Notch pathway is deregulated in different cancers, and it is believed that Notch regulates the formation of cancer stem cells and the initiation of epithelial-mesenchymal transition phenotype.
Sonic hedgehog or hedgehog (Hh) signaling pathway was initially discovered in the fruit fly as a regulator of body segmentation in 1980. Hh signaling pathway forms one of the networks of major regulators of cell differentiation, proliferation, and cell polarity. This pathway is a fundamental part of embryonic development. It was shown that mutations generated in the Hedgehog pathway resulted in defective axial patterning, including cyclocephaly or holopronsencephaly, a cephalic disorder in which the Prosencephalon fails to develop in two hemispheres during embryogenesis. Finally, it was discovered that alterations in Hh signaling pathway were also associated with cancer development. Hh signaling has been shown to be crucial for the maintenance and expansion of CSCs. In other words, deregulation of Hh signaling has been linked with development of CSC formation and EMT development. This is seen in different types of tumors, especially in gastrointestinal cancers, leukemia, medulloblastoma, lung, and pancreatic cancer. Hh, like the Wnt signaling pathway, is one of the major regulators of cell differentiation and proliferation. It has been shown that inhibition of Hh signaling depresses self-renewal of pancreatic cancer stem cells and reverses chemoresistance. Specifically, they found that PANC-1 tumorsphere has properties of stemness and differentiation and is highly tumorigenic. All these pathways, in addition to their role in ESCs, SSCs, and CSCs self-renewal, have an important function as modulators of epithelial-mesenchymal transition initiation in cancer cells.
______
As is true for the rest of cancer genetics, information in this field is rapidly evolving. Large-scale bioinformatics and conceptual advances are integrating the CSC theory into the mainstream of cancer research and biology, as well as into the design for new diagnostic and therapeutic strategies. For example, it appears that much like hematopoietic stem cells, the CSC niche favors oligoclonality and some genetic diversity. Thus, clonal competition can ensue, giving rise to heterogeneous tumors and maintaining a reservoir of cells that can re-establish the tumor when a therapy effectively kills the predominant CSC clone and its progeny. Similarly, clonal competition can facilitate distant spread by selection of cells with different capabilities. An extreme example may be the potential for a single tumor cell (or a small population of oligoclonal cells in a tumor) to give rise to histologically distinct tumors—an event observed in xenotransplanted sarcomas.
______
______
Section-19
Cancer and immune system:
The immune system, including innate immune system and adaptive immune system, is composed of special factors, cells, tissues, and organs. It has been well known that the immune system can detect a wide variety of infectious organisms and other invaders, distinguish them from the own healthy cells and tissues, and prevent infections.
It has been documented that there is interaction between the immune system and cancer as evidenced by cancer immunosurveillance theory. Cancer immunosurveillance refers to the monitoring function of the immune system in cancer development. There are studies that have demonstrated that the immune system can recognize and eliminate aberrant cancer cells arising within the human body. Thus, the interaction between cancer and the immune system plays a pivotal role in cancer development. In cancer patients, the immune system is not sufficiently vigorous to eliminate cancer cells, suggesting that the antitumor immune system is suppressed. For instance, transplant recipients under continued immunosuppression displayed a significantly higher risk of developing de novo tumors such as lung cancer and noticeably increased risk for cervical cancer in immunosuppressed women.
The immune system interacts closely with tumor cells over entire stages of cancer progression from tumor initiation and development to metastasis, facilitating either cancer inhibition or promotion. The balance of these actions determines the eventual outcomes, which, in cases of clinically poor outcomes, are the consequences of immune evasion by tumors. The tumor microenvironment (TME) comprises not only tumor cells, but also immune cells and the surrounding stromal cells. Interestingly, cancer cells can take advantage of these immune cells to help them escape the host’s immune system. In addition, the movement of different subsets of immune cells into the TME is orchestrated by the chemokine system, followed by the regulation of tumor immune responses in a spatiotemporal manner, and cancer-related inflammation. Chemokines can also directly impact tumor cells and endothelial cells in the TME to regulate tumor cell growth and proliferation, angiogenesis, cancer stem-like cell properties, invasiveness, and metastasis.
It has been shown that several factors may contribute to antitumor immunosuppression, for instance, a low frequency of high-avidity antitumor T cells, presence of CD4+CD25+ regulatory T (Treg) cells, and various strategies of cellular-mediated tumor-induced immune evasion. Antitumor immunosuppression may also result from soluble factors and altered antigenicity. For instance, tumor-derived interleukin (IL)-18 induced immunosuppression in NK cell-controlled cancers. IL-1α upregulated TGF-beta in mesenchymal stem cells and thus induced immunosuppression, leading to the growth of prostate cancer cells. Therefore, immunological methods that eliminate antitumor immunosuppression and/or increase antitumor immunity can be very useful in the treatment of cancer.
_
The link between the immune system and cancer has been widely appreciated for over a century and was first highlighted by Rudolph Virchow over 150 years ago. The underlying basis for this relationship between cancer and immunity involves three basic principles of how the immune system acts to defend and protect an individual: it detects “nonself” antigens from pathogens or infected/malignant cells; it encompasses effector functions to specifically target and destroy the pathogen or infected/malignant cells while protecting the host; and it develops immunological memory via the adaptive immune responses for subsequent defense mechanisms following an injury or an attack against the host. Through this process, the immune system has acquired characteristics that give rise to the paradigm known as immunoediting, which provides a balance between immune surveillance and cancer progression in the realm of oncology. This multifaceted mechanism consists of the three primary phases: elimination, equilibrium, and escape, that contribute to cancer elimination, dormancy, and progression, respectively. Interestingly, this ability of cancers to evade or escape the immune response is now recognized to be one of the most distinguished cancer hallmarks, which provides the platform for treatments within the context of immunotherapies.
_______
_______
Overview of the immune system:
The immune system is comprised of several types of soluble bioactive molecules, cytokines, proteins, and cells that collectively form the multifaceted network of biochemical processes that recognize and defend against “nonself” proteins or antigens. To provide protection and maintain the host’s normal state of homeostasis, the immune system consists of two forms of immune responses: innate and adaptive (Figure below), Nonspecific and immediate immune responses are classified as innate due to their fast-acting nonspecific response against foreign antigens such as pathogenic microbes, allergenic antigens, or non-self-proteins or molecules (Figure below). While innate immunity is short-lived and not able to form an immunological memory, it is still able to distinguish between “self” and “nonself” or different groups of pathogens via receptors such as toll-like receptors (TLRs) and others that recognize specific danger associated or pathogen associated molecular patterns (DAMPs or PAMPs). For example, TLR7 is an intracellular receptor that can recognize single stranded RNA but it can also suppress induction of T regulatory cells (Tregs) which is helpful in the tumor environment. Other mechanisms by which the innate immunity imparts immediate protection to the host involve soluble bioactive proteins such as cytokines, chemokines and complement proteins. Notably, the cytokines have various functions depending on the microenvironment they were secreted in, the cells they were secreted by, the location of the receptor it binds to, and the signaling pathways that are activated following their binding to the receptor. On the other hand, while the complement proteins have three major signaling pathways that they are activated by (classical, alternative, and the lectin pathways), all pathways result in activated complement proteins. Upon activation, complement proteins function in opsonization, act as chemoattractant for other immune cells, and mediate cell/pathogen death by formation of membrane attack complex for lysis.
_
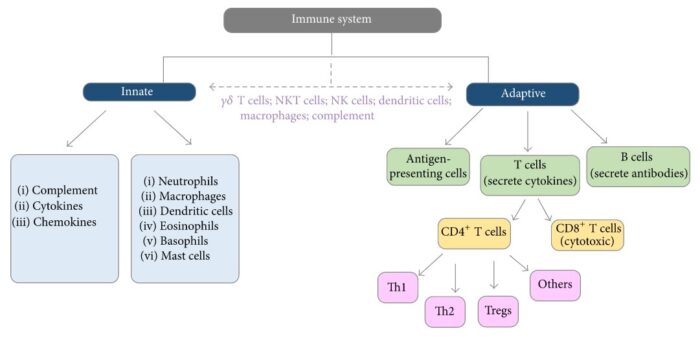
Figure above shows overview of the immune system: innate and adaptive immunity. An evolutionary bridge between both forms of immunity is observed due to the presence of γδ T cells, NKT cells, NK cells, dendritic cells, macrophages, and complement proteins. The innate immune responses include cells and soluble components that are nonspecific, fast-acting, and first responders in inflammation. In contrast, adaptive immunity encompasses immune components that are more specific for targeted antigens and capable of forming immunological memory.
_
The key players in cell-mediated innate immune responses involve phagocytes and natural killer (NK cells). These phagocytes (neutrophils, monocytes, and macrophages) facilitate immediate host protection by engulfing cells that express non-self-antigens or altered self-antigens and killing them with lysosomal enzymes. On the other hand, NK cells confer immune protection using major histocompatibility complex I (MHC class I) proteins which are universally expressed on cell surface of all nucleated cells. These NK cells secrete perforin and granzyme to induce apoptosis of cells that have abnormal or altered MHC class I expression if the cell has been compromised or a pathogen is expressed. Other cells such as eosinophils, basophils, and mast cells that release inflammatory mediators like chemotactic leukotrienes also contribute to the cellular innate immunity by recruiting more immune cells to the inflammation/injured site (Figure above). It is important to note here that, in humans, proteins known as human leukocyte antigens (HLA) are the equivalent to the MHC found in most vertebrates. Similar to NK cells, there are also cells known as NKT cells which possess qualities of both the NK and the T cells.
_
Contrary to the innate immune response, the adaptive immunity involves the development of immunological memory due to specific forms of immune responses targeting the antigens (Figure above). This form of immunity occurs over time and is not characterized as being a rapid response due to naïve lymphocytes, such as the T and B cells, gaining the ability to differentiate and mature into either effector T cells or antibody-secreting B cells (plasma cells) (Figure above). There are two types of T cells present in the immune system that are distinguished by their T cell receptor type: αβ T cells and γδ T cells. Only a small subset of T cells are classified as γδ T cells and can recognize “nonself” molecules by pattern recognition, thereby not requiring MHC-mediated antigen presentation. On the other hand, αβ T cells are further broken down into two other subsets known as CD4+ T cells and CD8+ T cells. Maturation of naïve CD4+ T cells to effector CD4+ T cells involves costimulation between major histocompatibility complex II (MHC class II) which are only present on antigen-presenting cells (B cells, macrophages, and dendritic cells) and T cell receptor on the naïve CD4+ T cells. Depending on the cytokine milieu present in the microenvironment and the presence of certain transcription factors during the time at which the costimulatory signal occurs, the CD4+ T cells can differentiate into several subsets of effector T cells such as T helper 1 (Th1) cells, T helper 2 (Th2) cells, or Tregs. Each of these subsets of CD4+ T effector cells can produce and secrete certain cytokines that modulate immune response accordingly. For instance, Th1 cells produce IFN-γ and interleukin-2 (IL-2) and play a role in autoimmunity. Notably, Th2 cells produce interleukins 4, 5, 10, 13, and 31 (IL-4, IL-5, IL-10, IL-13, and IL-31) and regulate immune responses involving extracellular pathogens as well as allergic diseases. Tregs, on the other hand, help reduce inflammation via production of transforming growth factor-beta (TGF-β), interleukin-35 (IL-35), and IL-10. Similar to NK cells in innate immunity, naïve CD8+ T cells rely on MHC class I for maturation into effector cytotoxic T cells. MHC class I molecules present on nucleated cells can recognize and bind to peptides derived from nonself antigens and altered or abnormal self-antigens. CD8+ T cells via the specific T cell receptor bind to the antigen/MHC class I complexes on the antigen-presenting cells (i.e., target cells) resulting in release of perforin and granzymes from CD8+ T cells and death of the target cell. Both types of T cells (CD4+ and CD8+) express other cell surface markers such as CD28 and CTLA-4 that participate in activating or inhibiting the naïve T cells, respectively, by binding to CD80/CD86 on antigen-presenting cells during costimulatory signaling.
_
Immune Checkpoints:
An important part of the immune system is its ability to tell between normal cells in the body and those it sees as “foreign” (such as germs and cancer cells). This allows the immune system to attack the foreign cells while leaving normal cells alone. Part of how the immune system does this is by using “checkpoint” proteins on immune cells. The checkpoints act like switches that need to be turned on (or off) to start an immune response. But cancer cells sometimes find ways to use these checkpoints to avoid being attacked by the immune system.
PD-1 is a checkpoint protein on immune cells called T cells. It normally acts as a type of “off switch” that helps keep the T cells from attacking other cells in the body. It does this when it attaches to PD-L1, a protein on some normal (and cancer) cells. When PD-1 binds to PD-L1, it basically tells the T cell to leave the other cell alone. Some cancer cells have large amounts of PD-L1, which helps them hide from an immune attack.
CTLA-4 is another checkpoint protein on some T cells that acts as a type of “off switch” to help keep the immune system in check. LAG-3 is another checkpoint protein on some types of immune cells that normally acts as a type of “off switch” to help keep the immune system in check.
_
For maturation and activation of B cells, antibody-secreting effector functions can be activated by T helper cell-dependent and cell-independent mechanisms resulting in a wide range of antibodies which are specific for the type of immune response that is initiated. Antibodies are also commonly referred to as immunoglobulins (Ig). They all possess a fragment antibody binding (Fab) domain that can bind to numerous antigens and a fragment crystallizable (Fc) domain that can bind to its corresponding Fc receptors on effector cells to mediate effector functions such as antibody-dependent complement cytotoxicity (ADCC). Whereas all naïve B cells express membrane-bound IgD and IgM, various other antibody isotypes such as IgA, IgG, and IgE are also produced by immediate and long-term memory plasma cells through immunoglobulin class switching, affinity maturation, and somatic hypermutations. Among these isotypes, certain antibodies such as IgA and IgG also have different subsets that they can be classified as isotypes. The various antibody isotypes and subsets can have distinct functions that they carry out in different conditions. However, in general, antibodies function to neutralize the antigen by binding to it, initiating ADCC, interacting with components of the classical complement pathway to induce complement dependent cytotoxicity (CDC), and also binding to antibody receptors on specific cells to activate their effector functions.
_
Similar to NK cells present in innate immunity, the adaptive immune responses also have their own version of NK cells known as NKT cells. These cells possess qualities of both the NK and the T cells, because they express NK 1.1, a natural killer cell-associated surface marker, along with the expression of T cell receptors (TCRs). However, some of their TCRs can differ from the normal TCR, thus classifying them as invariant NKT cells. These NKT cells are able to identify and bind to self or nonself lipids/glycolipids through the expression of CD1d molecule on antigen-presenting cells after which they secrete several cytokines such as IL-12 and IFN-γ for activation of other immune response.
_
While innate and adaptive immune responses have distinct characteristics that distinguish them from one another, both forms of immunity work in tandem. The innate immune response is initiated as the first line of defense prior to adaptive immunity. However, if the innate immune responses become overwhelmed by the antigens and are not sufficient for their clearance, then the onset of adaptive immunity occurs to aid in antigen removal and form immunological memory for subsequent exposures. Some of the components that serve as linker between innate and adaptive immunity include dendritic cells, macrophages, and complement protein. Dendritic cell and macrophages can both function in innate immunity by phagocytosing pathogens or innate immunity, but they also contribute to adaptive immune responses by serving as antigen-presenting cells for T cell activation. The role of complement proteins in innate immunity reflects its ability to eradicate cancer cells by formation of the membrane attack complex or by CDC. Moreover, in adaptive immune responses, the complement system can contribute to B cell activation by lowering the activation threshold and by mediating T cell activation and differentiation. Other cells like NK cells, NKT cells, γδ T cells, macrophages, dendritic cells, and complement proteins serve as evidence for the evolutionary bridge between innate and adaptive immunity.
_____
_____
Tumor antigens:
Tumors may express tumor antigens that are recognized by the immune system and may induce an immune response. These tumor antigens are either TSA (Tumor-specific antigen) or TAA (Tumor-associated antigen).
Tumor-specific:
Tumor-specific antigens (TSA) are antigens that only occur in tumor cells. TSAs can be products of oncoviruses like E6 and E7 proteins of human papillomavirus, occurring in cervical carcinoma, or EBNA-1 protein of EBV, occurring in Burkitt’s lymphoma cells. Another example of TSAs are abnormal products of mutated oncogenes (e.g. Ras protein) and anti-oncogenes (e.g. p53).
Tumor-associated antigens:
Tumor-associated antigens (TAA) are present in healthy cells, but for some reason they also occur in tumor cells. However, they differ in quantity, place or time period of expression. Oncofetal antigens are tumor-associated antigens expressed by embryonic cells and by tumors. Examples of oncofetal antigens are AFP (α-fetoprotein), produced by hepatocellular carcinoma, or CEA (carcinoembryonic antigen), occurring in ovarian and colon cancer. More tumor-associated antigens are HER2/neu, EGFR or MAGE-1.
_
Initially tumor antigens were broadly classified into two categories based on their pattern of expression: Tumor-Specific Antigens (TSA), which are present only on tumor cells and not on any other cell and Tumor-Associated Antigens (TAA), which are present on some tumor cells and also some normal cells.
This classification, however, is imperfect because many antigens thought to be tumor-specific turned out to be expressed on some normal cells as well. The modern classification of tumor antigens is based on their molecular structure and source. Table below provides a categorization of tumor antigens based largely on molecular criteria, but one could also consider categorizing these antigens based on their functional impact in the cancer setting.
General Categories and Examples of Tumor Antigens:
|
Category |
Example Antigen |
Cancer Histology |
|
Oncofetal |
CEA |
Colorectal carcinoma |
|
Immature laminin receptor |
RCC |
|
|
TAG-72 |
Prostate carcinoma |
|
|
Oncoviral |
HPV E6, E7 |
Cervical carcinoma |
|
Overexpressed/accumulated |
BING-4 |
Melanoma |
|
Calcium-activated chloride channel 2 |
Lung carcinoma |
|
|
Cyclin-B1 |
Multi |
|
|
9D7 |
RCC |
|
|
Ep-CAM |
Breast carcinoma |
|
|
EphA3 |
Multi |
|
|
Her2/neu |
Multi |
|
|
Telomerase |
Multi |
|
|
Mesothelin |
Ductal pancreatic carcinoma |
|
|
SAP-1 |
Colorectal carcinoma |
|
|
Survivin |
Multi |
|
|
Cancer-Testis |
BAGE family |
Multi |
|
CAGE family |
Multi |
|
|
GAGE family |
Multi |
|
|
MAGE family |
Multi |
|
|
SAGE family |
Multi |
|
|
XAGE family |
Multi |
|
|
CT9, CT10 |
Multi |
|
|
NY-ESO-1/LAGE-1 |
Multi |
|
|
PRAME |
Multi |
|
|
SSX-2 |
Melanoma, Multi |
|
|
Lineage Restricted |
Melan-A/MART-1 |
Melanoma |
|
Gp100/pmel17 |
Melanoma |
|
|
Tyrosinase |
Melanoma |
|
|
TRP-1/-2 |
Melanoma |
|
|
P.polypeptide |
Melanoma |
|
|
MC1R |
Melanoma |
|
|
Prostate-pecific antigen |
Prostate |
|
|
Mutated |
β-catenin |
Melanoma, Prostate, HCC |
|
BRCA1/2 |
Breast, ovarian carcinoma |
|
|
CDK4 |
Multi |
|
|
CML66 |
CML |
|
|
Fibronectin |
Multi |
|
|
MART-2 |
Melanoma |
|
|
p53 |
Multi |
|
|
Ras |
Multi |
|
|
TGF-βRII |
Colorectal carcinoma |
|
|
Posttranslationally altered |
MUC1 |
Ductal carcinoma, RCC |
|
Idiotypic |
Ig, TCR |
B, T leukemia, lymphoma, myeloma |
BRCA = breast cancer antigen; CDK4 = cyclin-dependent kinase-4; CEA = carcino-embryonic antigen; CML66 = chronic myelogenous leukemia (antigen) 66; CT= cancer testis; HPV = human papilloma virus; Ep-CAM = epithelial cell adhesion molecule; Ig = immunoglobulin; MART-1/-2 = melanoma antigen recognized by T cells-1/-2; MC1R = melanocortin-1-receptor; SAP-1 = stomach cancer- associated protein tyrosine phosphatase-1; TAG-72 = tumor antigen-72; TCR = T cell receptor; TGF-βRII = transforming growth factor-β receptor II; TRP = tyrosinase-related protein.
_
With the exception of oncoviral antigens such as the human papilloma virus E6 and E7 proteins (in cervical carcinoma), few tumor antigens would be considered as causal or required for maintenance of cellular transformation. Many of the tumor antigens cited in Table above, however, provide survival or dissemination benefits to developing cancers and could be considered as facilitators of disease progression. For instance, mutations in p53 and overexpression of cyclins (such as cyclin B1) allows for dysregulated growth; overexpression of antiapoptotic proteins such as survivin promotes tumor survival; mutation in fibronectin or posttranslational alteration in the MUC1 glycoprotein enhance the metastatic potential of tumor cells; and secreted tumor antigens, such as gangliosides, can inhibit immune function. Hence, there are multiple manners in which the same set of immunologically recognized tumor antigens may be dissected and classified.
_____
_____
Your immune system uses the same method to attack cancer as it uses against pathogenic microorganisms, but the process is more complicated because cancer cells are created by the body. Because of this, the normal ways to find and fight invading cells from outside the body aren’t always effective. If the body can’t tell the difference between tumor cells and normal cells, the tumor cells may be able to “hide” from the immune system. In some cases, the DNA changes (mutations) that cause the cancer may be different enough to stimulate an immune response similar to the response described for invading virus cells. If the immune system detects the cancer, the APCs show cancer cell materials to T cells, the primary players in the fight against cancer as seen in the figure below.
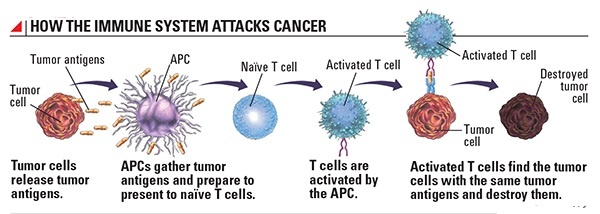
The MHC on APCs must connect to receptors on T cells, and the T cells must receive both Signal 1 and Signal 2 in order to become activated and multiply. If Signal 2 is not received, the response will shut down. A T cell can function properly against the cancer only if it recognizes the cancer as harmful, receives the proper signals to become activated, and continues to get signals to continue the attack.
Tumor cells can create cytokines, which means that cancer cells can communicate with and confuse other immune cells, allowing the cancer to take control of certain parts of the process that the body uses to regulate the immune response. So, even if the immune system recognizes the cancer, it may not be able to successfully start or maintain an attack long enough to kill the cancer cells.
The ability of T cells to become activated and attack cancer is at the heart of immunotherapy research. One specific area of research focuses on how cancer cells can trick the immune system into turning on “checkpoint pathways” early. Checkpoint pathways are part of the system of checks and balances that allow the immune cells to evaluate the attack against the threat at multiple stages. The pathways essentially function as the “brakes” when the body determines the response is no longer needed. By using signals to confuse other immune cells into putting on the brakes, the cancer can shut down the attack before it has responded effectively and thus, the cancer cells continue to grow. Blocking the effect of these checkpoint pathways can restore the normal function of the immune cells.
The longer the cancer cells face a weakened immune response, the more they’re able to adapt, and the easier it is for them to manipulate immune cells inside the tumor’s location (sometimes called the microenvironment area). The area, typically contains cancer cells, normal connective tissues that form the structure of the tumor, access to blood vessels that drive tumor growth and several cell types that contribute to tumor development. Immune cells found in this area are often referred to as tumor-infiltrating lymphocytes (TILs). Because the tumor can control cells in this area, the tumor can trick TILs into becoming useless or even helping the tumor grow. For example, APCs may be confused by signals from tumor cells, preventing the APCs from functioning properly, and making them incapable of sounding the alarm about a threat.
In some cases, tumors can upregulate (increase) the activity of regulatory T cells inside the area. With this increased activity, regulatory T cells are actually working to reduce the immune response around the tumor by turning off the other cancer-fighting T cells. It’s as if the tumor recruits the body’s own immune cells to fight off the attack, using the very processes that normally protect the body. The longer the immune system is exposed to the tumor, the weaker the immune response becomes.
______
______
Overview of relationship between immunity and cancer:
The ecological niche in which cell transformation and tumour progression occur is an essential component of malignancy. Innate and adaptive immunity play key roles in the tumour microenvironment (TME) by interacting with cancer cells as well as with stroma and the vascular bed. Immunity in all its diversity and plasticity acts as a double-edged sword during carcinogenesis, invasion, and metastasis. Appropriately activated T cells and innate immune effectors (natural killer [NK] cells) mediate early elimination of transformed cells and limit progression. In contrast, inflammatory cells and myeloid cells – in particular, macrophages – act as “corrupted policemen”, promoting carcinogenesis and tumour progression at different levels, including suppression of effective adaptive immune responses.
_
Inflammation, innate immunity, and cancer:
A connection between inflammation and cancer has long been perceived. Inflammatory cells including macrophages, neutrophils, mast cells, and eosinophils are present in the TME. Tumour-associated macrophages (TAMs) are prototypic inflammatory cells, playing a key role in the orchestration of the TME. Mononuclear phagocytes are extremely plastic. In the context of interferon-driven type 1 immune responses, macrophages acquire tumoricidal activity. Type 1 immunity signatures are generally associated with better prognosis in human tumours. Moreover, type 1 immunity resulting in M1 polarization of macrophages mediates the initial (elimination) phase in the natural history of carcinogenesis.
_
There are different types of tumor-associated-macrophages (TAMs), with pro- or anti-tumor activity. TAMs can be recruited into the primary tumor by cancer cell derived chemokines and cytokines (e.g. CSF1, VEGFA, CXCL2, CXCL12). TAM1 macrophages are inflammatory and generally thought to be tumor suppressive. Conversely, TAM2 macrophages can decrease CD8+ T cell infiltration and are typically pro-tumorigenic. A similar effect can be mediated by transforming growth factor (TGF)-β polarized tumor-associated-neutrophils (TANs). Both TAMs and TANs are thought to promote migration and intravasation of cancer cells.
_
During neoplastic progression, macrophage function is skewed in a pro-tumour direction (TAM2). Signals responsible for the protumour function of TAMs are known to originate from tumour cells (e.g. interleukin-10 [IL-10], transforming growth factor β [TGF-β]); T helper type 2 (Th2) cells, eosinophils, or basophils (IL-4 or IL-13, resulting in M2 activation); B cells (antibodies, immune complexes); and stromal cells (IL-1). There is evidence suggesting that the relative importance of different pathways for regulating the function of TAMs varies in different tissues. Single-cell analysis has added a new dimension to the dissection of myeloid cell diversity in cancer. Clusters of more than 10 differentiation/activation states have been identified. The microanatomical signals responsible for the diversity of cancer-associated myeloid cells remain to be defined.
_
Phagocytosis is the eponymous function of mononuclear phagocytes. CD47 on normal and tumour cells delivers a “don’t eat me” signal via signal regulatory protein 1α (SIRP1α) on macrophages. CD47 is amplified downstream of the oncogene MYC. CD47, which is one of the negative regulators (checkpoints) of myeloid cells, can serve as a therapeutic target. Recent evidence suggests that blocking CD47 may unleash antibody-dependent cellular cytotoxicity and phagocytosis mediated by TAMs. TAMs and other myeloid cells – for example, operationally defined myeloid-derived suppressor cells (MDSCs) – have now been shown to have impacts on diverse aspects of cancer progression, including tumour cell proliferation and invasion, construction of a metastatic niche, angiogenesis, and immunosuppression. Immunosuppression, a key function of myeloid cells, is discussed below.
_
Components of the humoral arm of innate immunity have recently been recognized as important elements in the TME. The complement system can also act as a double-edged sword by mediating complement-dependent cytotoxicity in the presence of antibodies or, alternatively, by recruiting tumour promoting myeloid cells. The long pentraxin PTX3 was shown to act as an extrinsic oncosuppressor, which is epigenetically silenced in selected human tumours. PTX3 silencing unleashes complement-driven recruitment and functional orientation of TAM2, which is responsible for tumour promotion and increased genetic instability.
Cytokines are a key component of tumour-promoting inflammation. In particular, IL-1 has been shown to drive myeloid cell infiltration, generation of MDSCs, and angiogenesis. Recent evidence is consistent with IL-1 being an important driver of progression in human tumours. The Canakinumab Antiinflammatory Thrombosis Outcomes Study (CANTOS) was originally designed to assess the impact of an anti-IL-1β antibody (canakinumab) on atherosclerosis-related cardiovascular pathology. In more than 10000 patients, blocking of IL-1β was associated with reductions of more than 50% in the incidence of and mortality from lung cancer. These and other results provide a strong proof-of-principle rationale for targeting tumour-promoting inflammation in human tumours.
_
Anti-tumour immunity and immunosuppression:
T-cell-orchestrated type 1 immune responses mediate host resistance during the early phases of carcinogenesis. Moreover, in human tumours, the presence of T cells and type 1 immunity or interferon signatures is associated with better prognosis. Genomics has provided a more in-depth view of immune cell recognition of tumour-specific antigens, arising from mutations, or tumour-associated antigens, resulting from overexpression of normal cell genes. Evidence in mouse and human tumours has indicated that mutations and genetic instability represent the fundamental molecular basis for T-cell-dependent anti-tumour immunity. The intersection of genomics and the dissection of immunity is paving the way to personalized immunotherapy approaches.
_
Failure of effective immunity is associated with progression and the appearance of clinical cancer. In the Darwinian TME, tumour cell-centred and host cell centred mechanisms of immune evasion drive progression, invasion, and metastasis. Mechanisms of physical exclusion (e.g. extracellular matrix deposition), and selection of less immunogenic variants, can hamper effective recognition.
T-cell exhaustion is an effector T-cell-intrinsic mechanism for failure to mount an effective immune response. Single-cell genomic analysis has provided new vistas on the T-cell receptor repertoire and functional properties of tumour-infiltrating lymphocytes. Regulatory T cells (Treg cells) have long been associated with immunosuppression in cancer. Single-cell analysis has led to the identification of molecules expressed by infiltrating Treg cells. For instance, the IL-1 decoy receptor IL-1R2 was found to be expressed at very high levels in infiltrating Treg cells.
Whereas a Th1-orchestrated cytotoxic T-cell-mediated response has a protective function, Th2-polarized T cells and Th17 cells trigger tumour-promoting cascades. IL-4 and IL-13 produced by Th2 cells or by eosinophils elicit alternative M2 polarization of macrophages, which results in tumour promotion. Evidence suggests that this pathway plays a dominant role in carcinoma of the breast and in pancreatic ductal adenocarcinoma. Th17 cells activate a neutrophil-dependent pathway of immunity to extracellular pathogens, and neutrophils can contribute to myeloid cell-mediated tumour promotion.
_
Whereas a skewed, inappropriate response and exhaustion are important determinants of the failure of immunity to restrain cancer, active immunosuppression has emerged as a dominant mechanism of progression. Checkpoints are physiological mechanisms to restrain uncontrolled T-cell activation and tissue damage. Targeting of the programmed cell death 1 (PD-1)/ programmed death ligand 1 (PD-L1) axis and cytotoxic T-lymphocyte associated protein 4 (CTLA-4) has had an unprecedented impact on cancer treatment. A host of molecular brakes acting on T cells as well as other cell types have been identified, and these represent candidate therapeutic targets. Immunosuppression in the TME is orchestrated by tumour cells and/or by stromal cells, in particular myelomonocytic cells. Tumour cells produce immunosuppressive cytokines (IL-10, TGF-β) and express triggers of checkpoint blockade, such as PD-L1. PD-L1 gene amplification was found to occur in Hodgkin lymphoma, in which PDL1 is also prominently expressed by TAMs. In general, the relative contribution of tumour cells versus myeloid cells to PD-L1 expression in the TME varies considerably in different human cancer types.
_
Myelomonocytic cells at different stages of differentiation or activation have the capacity to strongly suppress T-cell-mediated responses. MDSCs are operationally defined as a mixed population of relatively immature myeloid cells with potent suppressive activity. Depending on the system examined among MDSCs, suppression was mediated by neutrophils or, more frequently, monocytes. Monocytic MDSCs differentiate into TAMs in the TME.
TAMs were found to exert immunosuppressive activity via diverse mechanisms. These include immunosuppressive cytokines (IL-10, TGF-β), triggers of checkpoint blockade (e.g. PD-L1), amino acid metabolism (arginase, tryptophan metabolites), and prostaglandins. Prostaglandins are particularly significant in view of the protective effect of aspirin on several human tumour types.
_
B cells and antibodies are part of the anti-tumour response. However, evidence suggests that B cells can contribute to tumour progression in certain epithelial tumours, such as prostate cancer. B-cell-mediated tumour promotion has been shown to involve different mechanisms, such as production of immunosuppressive cytokines (IL-10) and/or production of antibodies and formation of immune complexes that skew TAMs in an M2-like direction.
Adaptive T-cell-orchestrated immunity and its subversion are central in the control of carcinogenesis and progression. Recent results have shed new light on the long-overlooked role of innate lymphoid cells. NK cells are a population of innate lymphoid cells that has not been credited with playing a major role in resistance against solid tumour carcinogenesis. Evidence suggests that NK cells mediate resistance against haematopoietic neoplasms and restrain haematogenous dissemination of cancer cells. The differentiation and activity of NK cells are also controlled by negative regulators. Recently, novel NK cell checkpoints (e.g. IL-1R8) were identified, and unleashed NK cells were found to mediate resistance to carcinogenesis and metastasis at NK-cell-rich anatomical sites, such as the liver and the lung. Elucidation of the molecular mechanisms that regulate the function of NK cells and innate lymphoid cells may pave the way to therapeutic strategies that are complementary to the current checkpoint blockade.
_
In a nutshell:
Immunity is an essential component of the TME and a key determinant of metastasis. Inflammatory cells, in particular TAMs, pave the way to tissue invasion and intravasation and provide a nurturing microenvironment for metastasis, serving as a component of the cancer cell niche at distant sites. NK cells are innate lymphoid cells that have long been considered to play a role in resistance against haematogenous dissemination of cancer cells, in particular to the lungs. Tumour progression and escape are associated with immunosuppressive pathways in innate and adaptive anti-tumour responses, which include, among others, suppressive myeloid cells, activation of checkpoint blockade, and induction and recruitment of Treg cells.
Key points:
-Immune cells and mediators of innate and adaptive immunity are essential components of the tumour microenvironment.
-Innate and adaptive immunity in the tumour microenvironment are double-edged swords.
-Appropriately activated adaptive immune responses mediate resistance to carcinogenesis and progression. In contrast, cancer-related inflammation orchestrated by innate immunity, such as macrophages and the complement system, facilitates tumour progression via several mechanisms, including suppression of adaptive immune responses.
-Components of innate immunity drive tumour promoting inflammation. Macrophages promote tumour progression and immunosuppression.
-T cells eliminate and edit cancer cells. Checkpoints and other pathways of suppression restrain the anti-tumour activity of T cells, natural killer cells, and macrophages.
______
______
The six cell-intrinsic hallmarks of cancer and effects on immune system:
There are numerous molecular differences between cancer cells and healthy cells. These differences can be classified into six hallmarks of cancer. Table below depicts the impact of these cancer-specific hallmarks on the immune system.
Impact of cancer-cell-intrinsic hallmarks on the immune system:
|
Cancer hallmark |
Factor involved or characteristic |
Effect on immune response |
|
Self-sufficiency in growth signals |
IL-4 |
• Promotes differentiation into TH2 cells |
|
|
IL-6 |
• Inhibits inflammatory response, through aberrant JAK–STAT3 signalling |
|
|
IL-10 |
• Promotes differentiation into TH2 cells |
|
Insensitivity to antigrowth signals |
TGFβ resistance |
• Inhibits antigen presentation • Inhibits T-cell proliferation • Inhibits NK-cell cytotoxicity • Activates TRegcells |
|
Evasion of apoptosis |
BCL-2-family proteins |
• Tumour antigens |
|
|
MUC1 |
• Subverts DC function • Tumour antigen |
|
|
Survivin |
• Promotes T-cell apoptosis, through inducing CD95L expression by tumour cells • Tumour antigen |
|
|
Reduced caspase activation |
• Reduces tumour immunogenicity |
|
Limitless replicative potential |
Mutated p53 TERT |
• Tumour antigen • Tumour antigen |
|
Sustained angiogenesis |
COX2 |
• Promotes TH2-cell responses • Inhibits T-cell cytotoxicity |
|
|
VEGF |
• Inhibits T-cell activation • Inhibits NF-κB activation in DCs |
|
Tissue invasion and metastasis |
Reduced NECL2 expression |
• Inhibits NK-cell cytotoxicity, through CRTAM • Inhibits CD8+ T-cell IFNγ production, through CRTAM • Leads to loss of adherence at epithelial cell–cell junctions |
|
|
Altered lysosome trafficking |
• Increases immunogenicity, through increasing cell-surface HSP70 |
|
|
MMPs |
• Induce cleavage of CD25 • Activate TGFβ • Promote shedding of ICAM1 and ICAM2 |
BCL-2, B-cell lymphoma 2; CD95L, CD95 ligand; COX2, cyclooxygenase-2; CRTAM, MHC-class-I-restricted T-cell-associated molecule; DC, dendritic cell; HSP70, heat-shock protein 70; ICAM, intercellular adhesion molecule; IFNγ, interferon-γ; IL, interleukin; JAK, Janus kinase; MMP, matrix metalloproteinase; MUC1, mucin-1; NECL2, nectin-like 2; NF-κB, nuclear factor-κB; NK cell, natural killer cell; p53, tumour-suppressor protein p53; STAT3, signal transducer and activator of transcription 3; TERT, telomerase reverse transcriptase; TGFβ, transforming growth factor-β; TH2 cell, T helper 2 cell; TReg cell, CD4+CD25+ regulatory T cell; VEGF, vascular endothelial growth factor.
_______
_______
Cancer Immunosurveillance to Immunoediting:
Cancer immunoediting process is envisaged to consist of three phases: elimination, equilibrium, and escape. Elimination phase corresponds to the original concept of cancer immunosurveillance, whereby nascent tumour cells are successfully recognised and eliminated by the immune system, thus returning the tissues to their normal state of function. Tumour cells that elude the immunosurveillance phase will progress to the immune editing phase, called the equilibrium phase of advanced oncogenesis, where tumour expansion and metastasis are minimal (tumour dormancy) and usually occur without symptoms. In the equilibrium phase, the immune system may eventually eliminate all tumour cells leading to an outcome similar to the elimination phase. In a second scenario, the constant interaction of the immune system with tumours over a long period of time may actually “edit” or sculpt the phenotype of the developing tumour, resulting in the immunoselection of a tumour that has been shaped into a less-immunogenic state. Tumours that are no longer susceptible to immune attack then progress into the immunoediting process, termed “escape.” The emergence of clinical symptoms of cancer generally correlates with the escape stage. Tumour subverts the immune system, either directly through its nonimmunogenic phenotype, or indirectly through a variety of immunosuppressive mechanisms.
_
Activation of host immune system is a natural way for cancer patients to fight this disease. Several studies have shown that cells of the immune system can recognize and destroy tumor cells. The process through which the immune system carries out this function is known as immunosurveillance. As shown in Figure below, continuous immunosurveillance takes place in the body to help the immune system to deal with “rogue” or abnormal cells. The outcome of this response is regulated by a process known as immunoediting. Cancer immunoediting refers to the dual role played by the immune system in host protection and promotion of tumor growth. Cancer immunoediting consists of three phases, which are elimination, equilibrium and escape. If the immune system is appropriately activated, tumor growth can be inhibited and they can be destroyed. However, in some situations, the immune system may can promote tumor progression through chronic inflammation and/or suppression of anti-tumor immune responses.
_
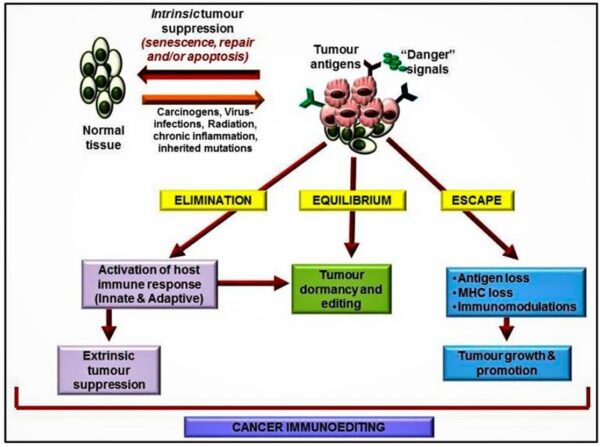
Figure above shows three phases of cancer immunoediting: elimination, equilibrium and escape.
During the elimination phase, the innate and adaptive arms of the host immune system work hand-in-hand to destroy cancer cells before these cells can be clinically detected. Many effector T-lymphocyte subsets and cytokines play key roles in eliminating tumor cells. Mechanisms by which cancer cell lysis occurs are via secretion of perforin from cytolytic immune cells (i.e., NK cells, NKT cells, γ T cells, and CD8+ T cells), ADCC, or CDC. Tumor cells that cannot be destroyed in the elimination phase can enter the equilibrium phase. The main role of the equilibrium phase is to prevent outgrowth of the tumor by enabling editing of tumor immunogenicity. In addition, T-helper-1 (Th1) cells as well as some of the cytokines that these cells produce (e.g., interleukin-12 (IL-12) and interferon-gamma (IFN-γ)) help to maintain tumor cells in a state of immune-mediated dormancy. However, maintaining immune cells constantly in this phase may allow emergence of unstable tumor cells that can overcome some of the barriers imposed by the anticancer immune responses. One of the reasons for this could be expression of new molecules on the tumor cells due to mutations, which are no longer recognized by the receptors of these lymphocytes. In addition, the tumor cells may secrete mediators that could induce an immunosuppressive state within the tumor microenvironment. When this happens, the tumor cells are no longer susceptible to the host immune system, enabling them to avoid the elimination and equilibrium phases and enter the escape phase. In the escape phase, tumor progression is no longer blocked by the host immune system and the tumor can be detected clinically. This escape phase is mediated through several immunosuppressive mechanisms one of which includes downregulation or aberrant expression of MHC class I on the cancer cell surface protecting it from cytotoxic effector functions of immune cells in the innate and adaptive immunity. Multiple mechanisms such as suppression of tumor antigen expression, induction of antiapoptotic pathways to prevent cytotoxicity, and cancer-induced immunosuppression aid in the escape of cancer cells from the elimination and equilibrium phases of immunity.
_
Figure below shows schematic representation of the immunoediting process consisting of three phases: elimination, equilibrium, and escape, termed the “three Es of cancer immunoediting. Carcinogenesis is envisaged as a multistep process resulting from cross talk of cancer-cell–intrinsic factors and host immune system (cell-extrinsic) effects.
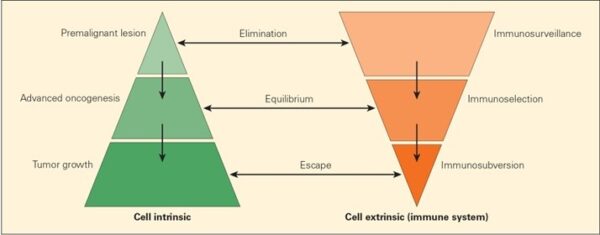
_
Immunoselection by cancer cells to escape/evade immune system:
Cancer immune escape is the process whereby tumor cells prevent their elimination by the immune system. Escape from immune system recognition often involves tumor-specific genomic alterations in immune-related pathways, a process named genetic immune escape (GIE). Tumours develop a series of strategies to escape/evade immune system, and these strategies are now recognized to be one of the most distinguished cancer hallmarks. These strategies are known as immunoselection.
- CD8+ cytotoxic T cells are a fundamental element of anti-tumor immunity. Their TCR receptors recognise antigens presented by MHC class I and when bound, the Tc cell triggers its cytotoxic activity. MHC I are present on the surface of all nucleated cells. However, some cancer cells lower their MHC I expression and avoid being detected by the cytotoxic T cells. This can be done by mutation of MHC I gene or by lowering the sensitivity to IFN-γ (which influences the surface expression of MHC I). Tumor cells also have defects in antigen presentation pathway, what leads into down-regulation of tumor antigen presentations. Defects are for example in transporter associated with antigen processing (TAP) or tapasin. On the other hand, a complete loss of MHC I is a trigger for NK cells. Tumor cells therefore maintain a low expression of MHC I.
- Another way to escape cytotoxic T cells is to stop expressing molecules essential for co-stimulation of cytotoxic T cells, such as CD80 or CD86.
- Tumor cells express molecules to induce apoptosis or to inhibit T lymphocytes:
-by expression of FasL on its surface, tumor cells may induce apoptosis of T lymphocytes by FasL-Fas interaction.
-by expression of PD-L1 on the surface of tumor cells leads to suppression of T lymphocytes by PD1-PD-L1 interaction.
- Tumor cells have gained resistance to effector mechanisms of NK and cytotoxic CD8+ T cell:
-by loss of gene expression or inhibition of apoptotic signal pathway molecules: APAF1, Caspase 8, Bcl-2-associated X protein (bax) and Bcl-2 homologous antagonist killer (bak).
-by induction of expression or overexpression of antiapoptotic molecules: Bcl-2, IAP or XIAP.
-by overexpression of the serine-protease inhibitor PI9 by tumour cells efficiently blocks the granzyme-B–perforin pathway of target-cell lysis.
-by downregulation or mutation of death receptors, methylation or mutation of the gene that encodes caspase-8, and overexpression of FLIP (caspase-8 (FLICE)-like inhibitory protein) or decoy receptors for TRAIL, all of which cause resistance to Cytotoxic T lymphocytes (CTLs) induced killing of tumour cells
- Tumours often downmodulate molecules that are involved in antigen processing and in antigen presentation by HLA class I molecules — including transporter associated with antigen processing 1 (TAP1), low-molecular-mass protein 2 (LMP2), LMP7 and tapasin — the expression of which is progressively lost during the development of colorectal carcinoma.
_
Immunosuppression and Tumor Progression:
Tumors can suppress immunity both systemically and in the microenvironment of the tumor. In addition to producing immunosuppressive molecules such as transforming growth factor β (TGF-β) and soluble Fas ligand, many human tumors produce the immunosuppressive enzyme indolamine-2,3-dioxygenase (IDO). This enzyme was previously known for its role in maternal tolerance to antigens from the fetus and, more recently, as a regulator of autoimmunity that mediates inhibition of T-cell activation. Stereoisomers of 1-methyl-tryptophan inhibit IDO, and when administered to tumor-bearing mice, they restore immunity and thereby allow immune rejection of the tumor. Such stereoisomers might have a role in the treatment of patients with cancer. The tumor microenvironment can be dominated by regulatory T cells that suppress antitumor effector T cells by producing the immunosuppressive cytokines TGF-β and interleukin-10. High numbers of these cells can be detected in non–small-cell lung cancer and ovarian cancer. Murine tumors that produce TGF-β can convert antitumor effector T cells into regulatory T cells, thereby escaping their own destruction by immune cells. The immunosuppressive effects of a tumor can also be systemic. An increase in regulatory T cells has been observed in the peripheral blood of patients with head and neck cancer or melanoma. Patients with colorectal cancer or pancreatic tumors have increased numbers of activated granulocytes and myeloid-derived suppressor cells, both of which suppress tumor-specific T cells in mice. Cancer can weaken the immune system by spreading into the bone marrow. The bone marrow makes blood cells that help to fight infection. This happens most often in leukaemia or lymphoma, but it can happen with other cancers too. The cancer can stop the bone marrow from making so many blood cells. A study in mice by UC San Francisco researchers has found that, depending on a cancer’s tissue of origin, tumors cause widespread and variable disruption of the immune system throughout the body, not just at the primary tumor site.
_____
_____
Immunodeficiency can lead to cancer:
Immunodeficiency, whether acquired in the case of human immunodeficiency virus (HIV) infection or congenital due to inborn errors of immunity (IEIs), presents clinically with not only infection and immune dysregulation but also increased risk of malignancy. The range of malignancies seen is relatively limited and attributable to the particular cellular and molecular defects in each disease. CD4+ T-cell lymphopenia in people living with HIV infection (PLWH) and certain IEIs drive the predisposition to aggressive B-cell non-Hodgkin lymphomas, including certain rare subtypes rarely seen in immunocompetent individuals. PLWH and IEI that lead to profound T-cell lymphopenia or dysfunction also are at risk of cancers related to oncogenic viruses such as Kaposi sarcoma herpesvirus, Epstein-Barr virus, human papillomavirus (HPV), and Merkel cell polyomavirus. IEIs that affect natural killer cell development and/or function heavily predispose to HPV-associated epithelial cancers. Defects in DNA repair pathways compromise T- and B-lymphocyte development during immune receptor rearrangement in addition to affecting hematopoietic and epithelial DNA damage responses, resulting in both hematologic and nonhematologic cancers.
_
The tipping point:
Medical research suggests that a strong immune system does protect against cancer. Immune system kills spontaneous blood cancer cells every day. A new study from Australia suggests B cells, a type of white blood cell, undergo spontaneous changes that could lead to cancer if the immune system does not carry out regular checks and kill them before they form tumors. Each and every one of us has spontaneous mutations in our immune B cells that occur as a result of their normal function. If cancerous B cells go on to form tumors they develop into B cell lymphomas, also known as non-Hodgkin’s lymphomas. The researchers found T cells of the immune system carry out regular checks to find cancerous and pre-cancerous B cells. This regular surveillance by the immune system is the reason why there are not as many cases of B cell lymphomas in the population, given how often the spontaneous changes occur.
If the immune system is so strong and sophisticated, why does it fail so often in fighting cancer?
The short answer: because the cancer overpowers it.
If we didn’t have an immune system, we’d all get cancer. There’s a fine balance between the burden of cell mutation and how well your immune system can fight it off. It is always alert to threats. It’s when the immune system is overwhelmed by a tumor that it fails to identify and respond to the threat. Indeed, it is possible, even likely, that your immune system may regularly fight off cancer or pre-cancer on a regular basis without you even knowing it. We all have a mechanism to filter out a small amount of cancer cells to prevent us from having visible cancer in the body. Over time, that balance becomes lost.
The tipping point at which cancer begins to overwhelm the immune system is not always known. Some of it has to do with the DNA of the tumor. Some of it has to do with the aggressiveness of the cancer. But research has shown that cancer cells exert tremendous sway over some innate and adaptive immune cells and recruit them to help cancer grow and travel. There is a very intricate balance in the immune system that is usually anti-tumorigenic, meaning it eliminates tumors, but in some cases, if this balance is altered, these cells may actually help tumors grow and develop into full-blown metastatic disease. An article in Nature describes how tumor-associated macrophages (TAMs) help breast tumors form. Macrophages are often found in large numbers in some breast tumors. While they are capable of attacking cancer cells, “macrophages within mammary tumors are inadvertently licensed to promote tumor growth and metastasis,” the Nature article says.
______
______
The Dark Side of IFN-γ: Its Role in Promoting Cancer Immunoevasion, a 2018 study:
Interferon-γ (IFN-γ) is a pleiotropic cytokine that has long been praised as an important effector molecule of anti-tumor immunity, capable of suppressing tumor growth through various mechanisms. On the contrary to such a bright side of IFN-γ, it has also been involved in promoting an outgrowth of tumor cells with immunoevasive phenotype suggesting an existence of a dark “tumor-promoting” side effect of IFN-γ. In this review, authors summarize this multi-functional role of IFN-γ in tumor context, how it promotes changes in tumor phenotype towards increased fitness for growth in immunocompetent host. Furthermore, authors summarize how IFN-γ is involved in homeostatic or cancer-triggered mechanisms to establish an immunosuppressive tumor microenvironment.
_____
_____
Mutated tumor suppressor genes block the body’s natural defense against malignancies, a 2021 study:
So-called tumor suppressor genes have long been known to block cell growth, preventing cancerous cells from spreading. Mutations in these genes, scientists believed, thus allow tumors to flourish unchecked. Now, Howard Hughes Medical Institute Investigator Stephen Elledge’s team has uncovered a surprising new action for many of these defective genes. More than 100 mutated tumor suppressor genes can prevent the immune system from spotting and destroying malignant cells in mice.
Conventional wisdom had suggested that, for the vast major of tumor suppressor genes, mutations allow cells to run amok, growing and dividing uncontrollably. But that explanation had some gaps. For example, mutated versions of many of these genes don’t actually cause rampant growth when put into cells in a petri dish. And scientists couldn’t explain why the immune system, which is normally highly proficient at attacking abnormal cells, doesn’t do more to nip new tumors in the bud.
Elledge’s new paper offers some answers. His team probed the effects of 7,500 genes, including genes known to be involved in human cancer. A third or more of those cancer-linked genes, when mutated, trigger mechanisms that prevent the immune system from rooting out tumors, often in a tissue-specific manner. These results reveal a fascinating and unexpected relationship between tumor suppressor genes and the immune system.
______
______
Stanford Medicine study shows how oncogene tricks immune cells, a 2023 study:
A novel Stanford School of Medicine study uncovers a direct link between a cancer-associated gene, Myc, and sugar patterns on cancer cell surfaces that tell immune cells to stand down.
Cancer-associated genes called oncogenes are well known to stimulate cell growth and division — causing tumors to balloon and spread. But now, researchers at the Stanford School of Medicine and Sarafan ChEM-H have found that one notorious oncogene called Myc also has a direct role in disguising growing cancers from the immune system. Myc is associated with more than 70% of human cancers, and blowing these cancers’ cover could lead to a new class of cancer therapy, the researchers believe. They found that a key component of the Myc-induced camouflage is a sugar molecule coating on the surface of cancer cells. This sugar sends a “stand down” signal to immune cells called macrophages that would normally engulf and destroy the cancer cells.
Myc drives the production of a glycosyltransferase called ST6GalNAc4, which is necessary to make a sugar molecule called disialyl-T that pops up in abundance on the surface of Myc-driven cancer cells. Disialyl-T binds to another molecule on the surface of macrophages, flipping them in one fell swoop from foe to friend.
This interaction is important; leukemia cells driven by Myc expression, but unable to make ST6GalNAc4, grew more slowly in laboratory mice and generated a stronger immune response to the tumor than cells that could make ST6GalNAc4. Additionally, researchers found that people with cancers that have high levels of both Myc and ST6GalNAc4 have a poorer prognosis and fewer immune cells near their tumors than those with lower levels.
_______
_______
Section-20
Cancer in animals:
Cancer is prevalent in the animal kingdom. Cancer affects mammals, reptiles, birds, fish, and mollusks. Some species develop cancers similar to humans. Examples of these species include pets in our homes such as cats, dogs and domestic animals such as horses. Approximately 1 in 4 dogs will, at some stage in their life, develop neoplasia. Almost half of dogs over the age of 10 will develop cancer. Dogs get cancer at roughly the same rate as humans, while there is less information about the rate of cancer in cats. Some cancers, such as lymphoma, are more common in cats than in dogs. Second-hand cigarette smoke is one of the risk factors for the development of cancer in domestic animals. Wild animals get cancers too. Sea animals can also get cancer. The risk of cancer in sea animals is increased by pollutants in water bodies. A population of sea lions in California is known to suffer from urogenital cancers. In St. Lawrence Estuary, Canada, 27% of beluga whales suffer from intestinal cancer. Sharks can get melanoma, a form of skin cancer. Animal species differ dramatically in their cancer rates and ages of disease onset. For example, 50 to 90% of aged mice die of cancer, while in humans this number is approximately 23%. The age of onset of cancer also varies greatly depending on the lifespan of the species. While it takes a mouse, on average, two years to develop cancer, it takes decades for long-lived species. Several species are known to be extremely cancer resistant. These include the naked mole rat, blind mole rat, elephant, bats and bowhead whale.
____
Peto’s Paradox:
Complex multicellular organisms are built of millions to quadrillions of cells, ultimately all being derived from a single cell, the zygote. During the course of the organisms’ lifetime and owing to various mutational processes, cell lineages tend to accumulate mutations. While the majority of mutations are harmless, some enable cells to escape cell cycle control, to grow and proliferate uncontrollably, resulting in cancer. Cancer is a multistage process, where a set of mutations is required for both initiation and malignant progression. Given that every cell division carries a risk of generating mutations, organisms with large bodies (composed of more cells) and extended longevities (with a longer time to accumulate mutations) should be more likely to develop cancer. Theory has it that the more the cells in an organism and the longer an organism lives, the more the likelihood of one of its cells succumbing to a random mutation and becoming cancerous. It would, therefore, be expected that larger organisms are more prone to cancers. Indeed, within humans and dogs, larger individuals are more likely to develop cancer than smaller ones. Similarly, increasing age is one of the most potent carcinogenic factors in species in which cancer aetiology is well studied. This correlation was disputed by Richard Peto, in what is referred to as the Peto’s paradox. While current evidence suggests that large body size and extended longevity result in increased cancer risk within species, this relationship may not hold across taxa.
Limited data available so far indicate that vertebrates do not face clear size-dependent cancer risks despite their size and longevity varying by orders of magnitude. This poses a logical challenge, first formulated Richard Peto. He noted that although mice have approximately 1,000 times fewer cells and >30 times shorter lifespans than humans, their risk of carcinogenesis is not markedly different (coined as Peto’s paradox). Richard Peto argues that the prevalence of cancer does not correlate with the number of cells in an organism. Peto’s paradox is demonstrated by animals such as the bowhead whale, elephant, and the naked rat mole. Peto’s paradox is an evolutionary conundrum that has puzzled the scientific community and has led to lively debate regarding the evolution of anticancer mechanisms. It is often postulated that natural selection on large size or extended longevity is inherently inseparable from the evolution of anticancer defences. Knowledge gained from investigating Peto’s paradox might thus largely contribute to our knowledge on natural anticancer mechanisms that could potentially be harnessed for medical use. Further, understanding cross-species variation of cancer vulnerability is an important next step in animal health and welfare.
____
____
Cancer resistance in some animals:
Cancer resistance has evolved multiple times in mammals. Species that display cancer resistance include the largest mammals such as whales and elephants, subterranean long-lived mammals (the naked mole rat and the blind mole rat), long-lived squirrels and bats. The specific mechanisms differ and were shaped by species ecology, lifestyle, and body characteristics. These mechanisms are beginning to be understood. The known mechanisms include duplications of the TP53 gene in elephants, overproduction of high molecular mass hyaluronan (HMM-HA) in the naked mole rat, interferon-mediated concerted cell death in the blind mole rat, and reduced growth hormone (GH)–insulin-like growth factor 1 (IGF1) signaling and microRNA (miRNA) changes in bats.
Bowhead whales are among the largest mammals. They can grow up to 20 meters long and weigh up to 100,000 kilograms. Bowhead whales have as estimated 3.7 quadrillion cells. With so many cells and such a long life expectancy, it would be expected that the Bowhead whales be prone to cancer. This is because the cells of bowhead whales have plenty of opportunities to mutate and become cancerous. However, Bowhead whales do not get cancer. Bowhead whale genome encodes a species-specific retroduplicated CDKN2CRTG gene that influence cellular phenotypes such as cell cycle progression and DNA damage repair in ways that are beneficial for aging and cancer resistance. Bowhead whales can live at least 211 years.
Naked rat mole produces hyaluronan, a polymeric molecule that prevents it from developing tumors. Hyaluronan is a thick sugary like substance, present in the spaces between cells. Even when these cells mutate, the hyaluronan prevents them from further division. Humans also produce hyaluronan. Though, the hyaluronan of humans is different from that of the naked rat mole. The hyaluronan of the naked rat mole is in longer chain form, whereas that of humans is in shorter chain form. Besides, naked rat moles have 5 times the level of hyaluronan as humans do. Naked rat moles can live for up to 30 years, whereas a similar sized mouse lives only up to 4 years.
Elephants have lower cancer rates. Only 5% of elephants die from cancer. The low cancer rates in elephants can be attributed to the fact that elephants have extra copies of the p53 gene, a tumor-suppressing gene. The p53 gene is the guardian of the genome. The p53 gene repairs damaged DNA by stopping the cell from proliferating so that it can repair itself. The gene also causes programmed cell death (apoptosis) of the cells that cannot be fixed.
Understanding how and why some animals rarely or do not get cancer at all could help us prevent or treat cancer. Scientists are investigating how this new knowledge could help develop cancer treatment. Scientists could create drugs that mimic the mechanism through which these animals resist cancer. One of the drugs being tested is Nutlin. In most human cancers, the p53 gene is mutated. Nutlin works by protecting the p53 gene, ensuring that it does not get destroyed. Other studies seek to determine whether the cancer-protective genes in these animals could be implanted into humans to protect against cancer, other age-related diseases and promote longevity. Other studies explore ways in which hyaluronan in humans can be regulated to simulate that in naked rat moles and such a treatment could be in the market soon, as hyaluronan is already used clinically. All that needs to be done is use the naked mole-rat long-chain version.
On the contrary, it is not clear whether these treatments will work well in humans. Molecules such as long-chain hyaluronan could have adverse effects on humans. Besides, genetic manipulation to incorporate these animals’ cancer-fighting genes in humans could result in incompatibility issues.
Several experiments using mice have demonstrated that the addition of numerous copies of the p53 gene in mice increased their resistance to cancer. However, the mice aged faster and were unable to produce offsprings at a younger age. Some of their internal organs shrunk. This gene in humans could have similar effects or worse. Genetic manipulation to treat or prevent cancer must be practiced with caution.
____
____
Cancer risk across mammals, a 2021 study:
Cancer is a ubiquitous disease of metazoans, predicted to disproportionately affect larger, long-lived organisms owing to their greater number of cell divisions, and thus increased probability of somatic mutations. While elevated cancer risk with larger body size and/or longevity has been documented within species, Peto’s paradox indicates the apparent lack of such an association among taxa. Yet, unequivocal empirical evidence for Peto’s paradox is lacking, stemming from the difficulty of estimating cancer risk in non-model species. Here authors build and analyse a database on cancer-related mortality using data on adult zoo mammals and map age-controlled cancer mortality to the mammalian tree of life. Authors demonstrate the universality and high frequency of oncogenic phenomena in mammals and reveal substantial differences in cancer mortality across major mammalian orders. Authors show that the phylogenetic distribution of cancer mortality is associated with diet, with carnivorous mammals (especially mammal-consuming ones) facing the highest cancer-related mortality. Moreover, they provide unequivocal evidence for the body size and longevity components of Peto’s paradox by showing that cancer mortality risk is largely independent of both body mass and adult life expectancy across species. These results highlight the key role of life-history evolution in shaping cancer resistance and provide major advancements in the quest for natural anticancer defences.
_____
_____
Moral of the story:
_
-1. Cancer is common worldwide, affecting about 1 in 3 people during their lifetime. Cancer is a disease of molecules and genes. Cancer refers to any one of a large number of diseases characterized by the development of abnormal cells that divide uncontrollably and have the ability to infiltrate and destroy normal body tissue, and often have the ability to spread throughout your body. Every case of cancer is unique, with its own set of genetic changes and traits. There are more than 200 different types of cancer.
_
-2. Human body consists of 100 trillion cells. Each day, the human body makes about 300 billion new cells. New cells are made to replace old/dead cells and for growth. Cells get old and die after a certain amount of time, and replication ensures that new cells are made to take their place. When they are acting normally, cells “know” which other cells to join up with and stick to – and they also know when to stop replicating and die. Each type of cell has a particular role and set of knowledge or instructions in their DNA (genes). New cells are made by process of cell division (e.g., mitosis). An estimated 1016 cell divisions take place in a normal human body in the course of a lifetime. Even in an environment that is free of mutagens, mutations will occur spontaneously at an estimated rate of about 10-6 mutations per gene per cell division—a value set by fundamental limitations on the accuracy of DNA replication and repair. Thus, in a lifetime, every single gene is likely to have undergone mutation on about 1010 separate occasions in any individual human being. Among the resulting mutant cells, one might expect that there would be many that have disturbances in genes that regulate cell division and that consequently disobey the normal restrictions on cell proliferation. From this point of view, the problem of cancer seems to be not why it occurs but why it occurs so infrequently. Clearly, if a single mutation were enough to convert a typical healthy cell into a cancer cell that proliferates without restraint, we would not be viable organisms. Many types of evidence indicate that the genesis of a cancer typically requires that several independent, rare accidents occur in the lineage of one cell. The development of a cancer requires mutations in many genes—perhaps ten or more.
_
-3. Because 300 billion new cells are made every day by process of cell division i.e., replication, there are inevitably going to be mistakes from time to time. A tumour occurs when there is a mistake in the copying process, and the replicated cell is faulty. The worst mistake is that they continue to replicate themselves without any limit. In addition, they are often unspecialised or undifferentiated, so they don’t serve a purpose, and they do not go through normal cell life cycles. At its heart, cancer is the result of uncontrolled growth of abnormal cells. Our bodies are composed of trillions of cells, all working together. In cancer, one of those cells stops paying attention to the normal signals that tell cells to grow, stop growing or even to die. Cancer cells still share many of the same needs and properties of normal cells but they become independent of the controls that make our body function smoothly. Cancer cells do not grow more quickly than normal cells, but they do not self-destruct and have no limits to how often they divide. They don’t know when to stop replicating and when to die. And they don’t always stick together, so they might break away and move through the blood vessels or lymphatic system and start growing somewhere else in the body. This process is called metastasis.
_
-4. Cancer is a genetic disease caused by accumulation of DNA mutations and epigenetic alterations leading to unrestrained proliferation of cells. Cancer cells and all of the cells of our body share roughly the same 25,000 genes. The difference between cancer cells and healthy cells is that cancer cells turn off genes that would regulate their growth, and they turn on genes that help them to grow or proliferate.
Cancer-related genetic changes occur because:
- random mistakes in DNA happen as cells multiply/replicate. The human genome has three billion base pairs per haploid set of chromosomes, and 6 billion base pairs are replicated during the S phase of the cell cycle in mitosis as each somatic cell has two copies of genome.
- DNA is altered by carcinogens in environment, such as chemicals in tobacco smoke, UV rays from the sun, and the human papillomavirus (HPV)
- DNA changes were inherited from parents
DNA changes, whether caused by a random mistake or by a carcinogen, can happen throughout our lives and even in the womb. While most genetic changes aren’t harmful on their own, an accumulation of genetic changes over many years can turn healthy cells into cancerous cells. The vast majority of cancers occur by chance as a result of this process over time.
_
-5. The billions of cells that make up a tumour are descended from a single cell, in which disturbance of the genetic and epigenetic codes caused remodeling of the control circuits that governed that cell’s existence. Cancer cells are created when the genes responsible for regulating cell division are damaged. Carcinogenesis is caused by mutation and epimutation of the genetic material of normal cells, which upsets the normal balance between proliferation and cell death. This results in uncontrolled cell division in the body. More than one mutation is necessary for carcinogenesis. In fact, a series of several mutations to certain classes of genes is usually required before a normal cell will transform into a cancer cell. The basic processes of proliferation (increase in cell number by cell division), differentiation (cell specialization into different tissue types), and apoptosis (programmed cell death) are guided by two innate programs in cells, the genetic code and the epigenetic code. In cancer each of these codes ultimately becomes altered regardless of whether the disease originated from an external or internal factor. Indeed, a fundamental characteristic of a tumour cell is that it begets a tumour cell. In other words, cancer, once manifest, becomes an inherited disease of the cell and is therefore self-perpetuating. The hereditary nature of cancer at the cellular level explains why alterations have been found in both the genetic and the epigenetic codes in tumour cells. The number of alterations seen in the coded programs increases as tumours progress to more advanced stages.
Tumor progression involves successive rounds of mutation and Natural Selection. At each stage, one cell acquires an additional mutation that gives it a selective advantage over its neighbors, making it better able to thrive in its environment—an environment that, inside a tumor, may be harsh, with low levels of oxygen, scarce nutrients, and the natural barriers to growth presented by the surrounding normal tissues. The offspring of this well-adapted cell will continue to divide, eventually taking over the tumor and becoming the dominant clone in the developing lesion. Thus, tumors grow in fits and starts, as additional advantageous mutations arise and the cells bearing them flourish.
_
-6. Cancer is a pathologic accumulation of clonally expanded cells derived from a common precursor. Cancer stem cell (CSC) hypothesis is based on the concept that tumors are hierarchically organized into a subpopulation of cells that retain or acquire the capacity for self-renewal and are capable and responsible for initiating and maintaining the tumor. Another subpopulation of cells that consists of the CSC progeny undergo partial to complete differentiation and lose the capability to support the tumor, albeit they still contribute to the morbidity of cancer. Although CSCs only constitute a low percentage of the total tumor mass, these cells can regrow the tumor mass on their own. The most critical properties of CSCs are their self-renewal ability and their capacity for differentiation into heterogeneous populations of cancer cells. This hypothesis fundamentally altered the way we understand cancer but also gave rise to a debate regarding how widely this model applies. The competing hypothesis is based on a model where all the cells in a tumor possess an equal capacity for self-renewal and is commonly referred to as the stochastic model that hypothesizes that each cell in a tumor mass has the potential for propagating tumor.
_
-7. Cancer stem cells (CSCs) is a sub-population of cells that share many common characteristics with somatic stem cells (SSCs). The lineages of cells in which all these DNA alterations accumulate are difficult to trace, but two recent lines of evidence suggest that normal stem cells may be the cells of origin in cancers.
- First, there exists a highly positive correlation between the risk of developing cancer in a tissue and the number of normal stem cell divisions taking place in that same tissue. The correlation applied to 31 cancer types and extended across five orders of magnitude. This strongly suggests that the main factor in cancer initiation is the fact that “normal” stem cells divide, which implies that cancer originates in normal, healthy stem cells.
- Second, statistics show that most human cancers are diagnosed in older people. A possible explanation is that cancers occur because cells accumulate damage through time. DNA is the only cellular component that can accumulate damage over the entire course of a life, and stem cells are the only cells that can transmit DNA from the zygote to cells late in life. Other cells, derived from stem cells, do not keep DNA from the beginning of life until a possible cancer occurs. This implies that most cancers arise from normal stem cells.
The above two lines of evidence show that healthy stem cell transforms into CSC due to mutations and epimutations. The strongest evidence for the CSC theory comes from studies in acute myelogenous leukemia (AML), when Bonnet and Dick performed serial transplantation experiments to show that only rare cells with high self-renewal capacity isolated from AML patients could initiate leukemia in immunocompromised murine models. Later the cell responsible for tumor initiation was identified by its phenotype as CD34+CD38−, a remarkably common phenotype of a normal hematopoietic stem cell (HSC). Additional studies have supported the idea of CSCs and currently this idea is becoming commonly accepted. While CSCs might represent a small fraction of the cells in the heterogeneous tumors, they likely play a fundamental role in cancer initiation, progression, and metastasis as well as in therapy failure. CSCs are also called tumor-initiating cells since they possess several important properties of SSCs: self-renewal, unlimited proliferation potential, slow replication, resistance to drugs, and the capacity to differentiate, giving rise to daughter cells, which make up the bulk of the tumor. While CSCs can initiate tumor formation, their daughter cells are incapable of forming new tumors by metastasis.
_
-8. Almost all cancers are believed to be clonal in the sense that all the cells of a given person’s cancer are descended from just one original somatic cell that underwent mutations and/or epimutations in cell division regulatory genes. The evidence includes such facts as that, in female mammals (where only one of the two X chromosomes is active in any given cell, with half the somatic cells having one of the Xs active and the other having the other X active) all the cells of any given tumor have the same active X. Likewise, cancers in chimeric animals (such as tetraparental mice) are found to contain only cells derived from one of the original embryos from which the animal was formed. In other words, cancer does not spread from cell to cell; the enlargement of tumors does not involve any kind of recruitment of previously normal cells; and it is not “catching”.
_
-9. Different from normal cells, cancer cells have various unique characteristics known as hallmarks of cancer. These characteristics included (1) self-sufficiency in growth signals, (2) insensitivity to antigrowth signals, (3) ability to evade apoptosis, (4) limitless replicative potential, (5) sustained angiogenesis, (6) capacity to invade tissues and metastasize, (7) abnormal metabolic pathways, (8) evading the immune system, (9) genome instability, and (10) inflammation. In principle, the cancer phenotypes proposed as hallmarks are based on the somatic mutation theory (SMT) of oncogenesis and its cell-centered variants that proposes that cancer originates when normal cells accumulate DNA mutations and/or epimutations over time and lose the ability to grow and proliferate in a regulated manner, leading to unrestricted cell proliferation. Others, though, argued that cancer is a tissue-level disease and cataloguing such cellular-level hallmarks are misleading.
_
-10. Cells divide in response to external signals that ‘tell’ them to enter the cell cycle. These signaling molecules bind to their target cells and send signals into the nucleus. The result is that the genes responsible for cell division are turned on and the cell divides. For example, a cut in the skin leads certain blood cells, platelets, to produce a growth factor that causes the skin cells to reproduce and fill the wound.
Somatic cells replicate and divide by the process of mitosis, in which genetic material is replicated and one parent cell divides into two ‘daughter’ cells. Cell division is a normal process that allows the replacement of dead cells or growth of tissues and organs. The cell division process occurs as an orderly progression through five different stages. These five stages are collectively known as the cell cycle. The cell cycle is an orderly sequence of events. Cells on the path to cell division proceed through a series of precisely timed and carefully regulated stages. The cell cycle is divided into five stages: G0, G1, S, G2 and M. ‘G’ stands for ‘gap’, ‘S’ represents ‘synthesis’, and ‘M’ stands for ‘mitosis’. Cell increases in size (G1 stage), copies its DNA (S stage), prepares to divide (G2 stage), and divides (M stage). The G0 phase is the resting phase. The genes that code for the positive cell-cycle regulators are called proto-oncogenes and negative cell-cycle regulators are called tumor suppressor genes. Proto-oncogenes control the growth and division of cells by coding for proteins that form a signaling “cascade.” This cascade relays messages from the exterior of the cell to the nucleus, where a molecular apparatus called the cell cycle clock resides. At the same time, tumour suppressor genes code for a similar cascade of inhibitory signals that also converge on the cell cycle clock. On the basis of the stimulatory and inhibitory messages it receives, the clock “decides” whether the cell should enter the cell cycle and divide. The cell cycle is controlled at three checkpoints. Integrity of the DNA is assessed at the G1 checkpoint. Proper chromosome duplication is assessed at the G2 checkpoint. Attachment of each kinetochore to a spindle fiber is assessed at the M checkpoint.
The cancer cell cycle uses the same mechanisms as a normal cell, but this process is highly unregulated. A cancer cell cycle would show checkpoint, growth, division, and cell death (apoptosis) anomalies. Two possible scenarios may lead to dysregulated cell growth and, ultimately, carcinogenesis: either a pathway that normally promotes cell growth may become disinhibited, or ‘turned on’, or a process which stops cell growth may become permanently disactivated. Also, when errors are found at the checkpoints, a normal cell will usually enter apoptosis and self-destruct. Cancer cell division, however, does not follow the rules of the regulatory checkpoints.
_
-11. Cancer is the result of unchecked cell division caused by a breakdown of the mechanisms regulating the cell cycle. The loss of control begins with a change in the DNA sequence of a gene that codes for one of the regulatory molecules. Faulty instructions lead to a protein that does not function as it should. Any disruption of the monitoring system can allow other mistakes to be passed on to the daughter cells. Each successive cell division will give rise to daughter cells with even more accumulated damage. Eventually, all checkpoints become nonfunctionally, and rapidly reproducing cells crowd out normal cells, resulting in tumorous growth.
Chemotherapy kills cells that are in the process of splitting into 2 new cells. Because cancer cells divide much more often than most normal cells, chemotherapy is much more likely to kill them. The fact that chemotherapy drugs kill dividing cells helps to explain why chemotherapy causes side effects. It affects healthy body tissues where the cells are constantly growing and dividing, such as: hair, bone marrow, skin etc. But normal cells can replace or repair the healthy cells that are damaged by chemotherapy. So the damage to healthy cells doesn’t usually last. Most side effects disappear once your treatment is over.
_
-12. Tissues which are typically lost and replaced at a high rate, such as epithelial cells, have a high mitotic index compared to non-dividing cells, such as muscle or nerve. This is one of the contributing factors as to why we see more cancers in rapidly-dividing cells: there is a greater likelihood of an error with each progression through the cell cycle. Another reason for higher cancer rates among epithelial cells is that these are more directly exposed to mutagenic environmental stimuli. A cancer case is estimated to be carcinoma 80 to 90% of the time. Carcinoma is cancer of epithelial cells.
Note:
Mitotic Index (MI) is the ratio between the number of cells in a population undergoing mitosis to the total number of cells in a population. Cancer cells in any tissue have an inappropriately higher mitotic index.
_
-13. Cancer cells are abnormal and unstable and show inter- and intra-tumor heterogeneity. Inter-tumor heterogeneity is manifested as the difference in tumor composition between different individuals for the same cancer type; while intra-tumor heterogeneity is described as differences between tumor cells inside of the same tumor. The heterogeneity of cancer cells introduces significant challenges in designing effective treatment strategies. Tumour heterogeneity has been observed in leukemias, breast, prostate, colon, brain, esophagus, head and neck, bladder and gynecological carcinomas, liposarcoma, and multiple myeloma.
_
-14. DNA damage is considered to be the primary cause of cancer. DNA damage leads to mutagenesis and mutagenesis leads to cancer. The primary structure of DNA is constantly subjected to alteration by cellular metabolites and exogenous DNA-damaging agents. These alterations may cause complex genetic changes, including deletions, fusions, translocations and aneuploidy, or, alternatively, single-base changes, which can be either transversions (change of purine to pyrimidine or vice versa) or transitions (change of purine to another purine or pyrimidine to another pyrimidine). Such alterations may ultimately lead to cellular death of unicellular organisms or degenerative changes and aging of multicellular organisms. DNA damage leads to mutation and epimutations of genes which leads to cancer. Exogenous and endogenous agents that induce DNA damage have been identified as major causes of many common cancers.
Every day, thousands of DNA damaging events take place in each cell of our body, but efficient DNA repair systems have evolved to prevent that. However, our DNA repair system and that of most other organisms are not perfect. In addition to mutations, which can be either inherited or somatically acquired, epigenetic silencing of DNA repair genes leads to accumulation of DNA damage that may promote tumorigenesis. Human cancers exhibit genomic instability and an increased mutation rate due to underlying defects in DNA repair genes. When expression of DNA repair genes is reduced, DNA damage accumulates in cells at a higher than normal rate, and this excess damage causes an increased frequency of mutation and/or epimutation.
_
-15. DNA is replicated with extreme fidelity in normal cells with a mutation rate of about 10-10 per base pair per cell division. Genome stability is a feature of every organism to preserve and faithfully transmit the genetic material from generation to generation or from one somatic cell to another. This includes an error-free replication of genetic material (DNA or RNA) and the repair of replication mistakes or damaged DNA/RNA.
In contrast, genome instability generally refers to the appearance of a high frequency of mutations in a genome, including point mutations, insertion/deletions, and large genetic rearrangements. Genomic instability is a characteristic of most cancer cells. It is an increased tendency of genome alteration during cell division. The great majority of human cancers show signs of a dramatically enhanced mutation rate: the cells are said to be genetically unstable. This instability can take various forms. Some cancer cells are defective in the ability to repair local DNA damage or to correct replication errors that affect individual nucleotides. These cells tend to accumulate more point mutations and small, localized DNA sequence changes than do normal cells. Other cancer cells have trouble maintaining the integrity of their chromosomes and thus display gross abnormalities in their karyotype.
Genetic instability would promote the formation of cancer, as it increases the probability that cells will experience a mutation that will lead toward malignancy. In fact, it seems that some degree of genetic instability may be essential for the development of cancer; at the very least it appears to make a powerful contribution to cancer progression.
Genomic instability arises from many different pathways, such as telomere damage, centrosome amplification, epigenetic modifications, and DNA damage from endogenous and exogenous sources, and can be perpetuating, or limiting, through the induction of mutations or aneuploidy, both enabling and catastrophic.
In normal cells, the intrinsic amount of genetic instability is low. Thus when normal cells hit a selection barrier—low levels of oxygen or a scarcity of proliferation signals, for example—they are very unlikely to be mutable enough to produce a cell that continues proliferating. In tumor cell precursors, an increased level of genetic instability makes it likely that at least one cell will contain the requisite genetic alteration to pass the selection barrier and continue the process of tumor progression. This genetic instability is retained in the lineage, and it can be measured in the resulting tumor. If the level of genetic instability is too high, many of the cells suffer deleterious mutations and either proliferate much more slowly than their neighbors or are eliminated by cell death. This excessive mutability can lead to extinction of the cell lineage.
_
-16. The discussion in above paragraphs proves that cancer is in fact an evolutionary cell growth with selection of those cells that are capable of surviving in adverse environment at the cost of useful functions. Survival of the fittest is the motto and not useful functions unless those functions aid in survival, so most cancer cells are dysfunctional but capable of surviving as compared to normal cells in neighborhood that perform useful functions but having lesser survival ability in adverse environment. So, cancer cells outgrow neighboring normal cells at the cost of useful functions. Remember, evolution favours survival and reproduction by on-going process of trial and error; both at macro level and at micro level. Cancer is microevolution of cells within eukaryotic multicellular organisms.
_
-17. Hereditary or germline mutations are gene mutations that are present at birth and exist in all of the cells of the body. Somatic or acquired mutations, in contrast, are those that occur after birth and are not passed down from one generation to another (not hereditary), and these mutations are not present in all cells. Somatic mutations are very common (about 1010 per gene in lifetime of a normal person); they are passed down only to the daughter cells of the cell undergoing the original mutation, and to their daughter’s daughter cells, etc. thus forming a clone of genetically different cells within the body. Nearly all somatic mutations are harmless, either with no effect or making the affected cells slightly abnormal (defective but not dangerous). Only certain mutations lead to cancer whereas the majority of mutations do not. According to the prevailing accepted theory of carcinogenesis, the somatic mutation theory, mutations in DNA and epimutations that lead to cancer disrupt orderly cell division process resulting in uncontrolled cell division of abnormal cells.
_
-18. Cancer is fundamentally a disease of regulation of tissue growth. In order for a normal cell to transform into a cancer cell, genes that regulate cell growth and differentiation must be altered. Genetic and epigenetic changes can occur at many levels, from gain or loss of entire chromosomes, to a mutation affecting a single DNA nucleotide, or to silencing or activating a microRNA that controls expression of 100 to 500 genes. In order for cells to start dividing uncontrollably, genes that regulate cell growth must be dysregulated. Proto-oncogenes are genes that promote cell growth and mitosis, whereas tumor suppressor genes discourage cell growth, or temporarily halt cell division to carry out DNA repair. Some classify DNA repair genes as subtype of tumor suppressor genes but others say that tumor-suppressor genes act to stop cell growth while DNA repair genes fix errors. Typically, a series of several mutations to these genes is required before a normal cell transforms into a cancer cell. Mutations and/or epimutations would transform proto-oncogene into oncogene and inactivate tumor suppressor gene. Unlike proto-oncogenes, which require that only one copy of the gene be mutated to disrupt gene function, both copies (or alleles) of a particular tumour suppressor gene must be altered to inactivate gene function. In many tumours one copy of a tumour suppressor gene is mutated, producing a gene product that cannot work properly, and the second copy is lost by allelic deletion. Most hematopoietic tumors and soft tissue sarcomas are initiated by activation of an oncogene, followed by alteration in tumor suppressor genes and other oncogenes. In contrast, most carcinomas are initiated by loss of function of tumor suppressor genes followed by alterations in oncogenes and additional tumor suppressor genes. Additional contributions to carcinogenesis come from genes that regulate programmed cell death (apoptosis) and genes involved in DNA repair. The average cancerous cell contains 60 or more mutations. The driver-versus-passenger concept was originally used to distinguish mutations that caused a selective growth advantage from those that did not. The mutations that confer a selective growth advantage to the tumor cell are called “driver” mutations.
_
-19. Solid tumors generally contain an abnormal number of chromosomes, a state known as aneuploidy; chromosomes from aneuploid tumors exhibit structural alterations such as translocations, deletions, and amplifications. These abnormalities reflect an underlying defect in cancer cells known as chromosomal instability and cancer cells become aneuploid only after many generations of clonal expansion. The molecular basis of aneuploidy remains incompletely understood. It is widely believed that defects in checkpoints, the quality-control mechanisms that halt the cell cycle if chromosomes are damaged or misaligned, contribute to chromosomal instability.
_
-20. Oncogenes were discovered through their presence in the genomes of retroviruses that are capable of causing cancers in chickens, mice, and rats. The nonmutated cellular homologues of these viral genes are called proto-oncogenes and are often targets of mutation or aberrant regulation in human cancer. In normal cellular environment, proto-oncogenes have crucial roles in cell proliferation and differentiation. Various forms of genetic mutation and alteration can convert a human proto-oncogene into an oncogene. Three main mechanisms have been identified: chromosomal translocation, gene amplification, and point mutation.
Point mutation (alternatively known as single nucleotide substitution) is a common mechanism of oncogene activation. Point mutations are also common mechanisms of inactivation of tumour suppressor genes. The restricted pattern of mutations observed in oncogenes compared to that of tumor-suppressor genes reflects the fact that gain-of-function mutations must occur at specific sites, while a broad variety of mutations can lead to loss of activity. Indeed, inactivation of a gene can in theory be accomplished through the introduction of a stop codon anywhere in the coding sequence, whereas activations require precise substitutions at residues that can somehow lead to an increase in the activity of the encoded protein under particular circumstances within the cell.
_
-21. Oncoproteins are the product (the proteins) that are coded for by oncogenes and are produced when the gene is transcribed and translated. There are many types of oncoproteins depending on the specific oncogene present, but most work to stimulate cell growth and division, inhibit cell death (apoptosis), or inhibit cellular differentiation (the process by which cells become unique). Oncoproteins can also affect other processes besides cell cycle, e.g., angiogenesis in tumor, metastasis etc. Oncoproteins include polypeptide growth factors, growth factor receptors, elements of intracellular signaling pathways, and transcription factors.
_
-22. The p53 (TP53) gene is the most commonly mutated gene in tumours. Practically every person who inherits a mutated copy of this tumour suppressor gene will develop some form of cancer. In particular, it prevents cells with damaged DNA from dividing or, when damage is too great, promotes apoptosis. Cells exposed to mutagens (chemicals or radiation capable of mutating the DNA) need time to repair any genetic damage they sustain so that they do not copy errors into the DNA of their daughter cells. When mutations occur, normal levels of the p53 protein rise, which slows the transition of the cell cycle from the G1 phase to the S phase. That extra time allows DNA repair mechanisms to effectively restore the DNA sequences to normal. The brakes on the cell cycle—high p53 levels—are then removed, and the cell proceeds to divide.
If there is a large amount of genetic damage, p53 triggers a series of biochemical reactions that cause the cell to self-destruct. Total functional inactivation of the p53 gene will cause genetic damage to accumulate in the cell and will also fail to set off apoptosis in severely injured cells.
In few types of cancer, inactivation of the p53 gene occurs not through mutation and loss of the alleles but through binding of the p53 protein with another protein (called an antagonist) that disables p53 function. One such antagonist, called MDM2, and overexpression of MDM2 in various tumours inhibits p53, therefore favouring uncontrolled cell proliferation. Other antagonists are the “early proteins” produced by cancer-causing strains of the human papillomavirus.
_
-23. Field defects are normal-appearing tissues with multiple alterations, and are common precursors to development of the disordered and over-proliferating clone of tissue in a cancer. Such field defects may have numerous mutations and epigenetic alterations. The somatic mutations and epigenetic alterations caused by DNA damage and deficiencies in DNA repair accumulate in field defects. A field defect probably arises by natural selection of a mutant or epigenetically altered cell among the stem and a mutant or epigenetically altered stem cell may replace the other nearby stem cells by natural selection. This may cause a patch of abnormal tissue to arise.
_
-24. The protein-coding DNA within the nucleus is about 1.5% of the total genomic DNA. Within this protein-coding DNA (called the exome), an average cancer of the breast or colon can have about 60 to 70 protein altering mutations, of which about 3 or 4 may be “driver” mutations, and the remaining ones may be “passenger” mutations. However, the average number of DNA sequence mutations in the entire genome (including non-protein-coding regions) within a breast cancer tissue sample is about 20,000. In an average melanoma tissue sample (melanomas have a higher exome mutation frequency) the total number of DNA sequence mutations is about 80,000. These high frequencies of mutations in the total nucleotide sequences within cancers suggest that often an early alteration in the field defect giving rise to a cancer is a deficiency in DNA repair.
_
-25. Breast cancer type 1 susceptibility protein is a protein that in humans is encoded by the BRCA1 gene. BRCA1 is a human tumor suppressor gene (also classified as DNA repair gene) and is responsible for repairing DNA. The human BRCA1 gene is located on the long (q) arm of chromosome 17 at region 2 band 1, from base pair 41,196,312 to base pair 41,277,500. BRCA1 and BRCA2 are unrelated proteins, but both are normally expressed in the cells of breast and other tissue, where they help repair damaged DNA, or destroy cells if DNA cannot be repaired. They are involved in the repair of chromosomal damage with an important role in the error-free repair of DNA double-strand breaks. If BRCA1 or BRCA2 itself is damaged by a BRCA mutation, damaged DNA is not repaired properly, and this increases the risk for breast and ovarian cancer. In one meta-analysis, cumulative breast and ovarian cancer risks for BRCA1 mutation carriers are 57 and 40%, respectively, while, for carriers of BRCA2 mutations, these risks are 49 and 18%, respectively. Furthermore, hereditary BRCA mutations have been also linked to pancreatic, prostate, and colon cancers. Of interest, germline mutations in BRCA1 versus BRCA2 associate with different subtypes of breast cancer. BRCA1 associated cancers are of the more aggressive triple negative subtype and appear at an earlier age than sporadic tumors. In contrast, BRCA2-associated tumors relate mostly with hormone receptor positive breast cancers.
_
-26. Different mutations in a single gene can predispose individuals to distinct cancer syndromes, whereas independent, single mutations of different genes can result in virtually the same disease, or at least diseases with indistinguishable phenotypes. This is not surprising when we consider that commonly affected genes are multifunctional and parts of complex, interactive networks or circuits. Thus, a mutation may only alter gene function along one biochemical pathway, leaving its interactions with other pathways intact. Moreover, mutations that contribute to most sporadic cancers are restricted to a small subset of genes, many of which also are associated with heritable cancer syndromes.
_
-27. Epigenetic events (epimutations) are those that can alter phenotype without changing the genotype. Epigenetics involves genetic control by factors other than an individual’s DNA sequence. Epigenetic changes can switch genes on or off and determine which proteins are transcribed. Many cancer risk factors, including ageing, inflammation, tobacco smoking, alcohol consumption, fungal toxins, biological agents, and diet as well as air and water pollution and certain endocrine disrupters, are associated with epigenome dysregulation. Almost all critical processes in cancer cells – such as self-sufficiency in growth signals, insensitivity to anti-growth signals, tissue invasion and metastasis, limitless replicative potential, sustained angiogenesis, and evasion of apoptosis – can be caused not only by genetic changes but also by epigenetic alterations. Silencing of some tumor suppressor genes in sporadic cancers occurs more frequently by epigenetic methylation than by mutation or deletion. These different mechanisms of gene silencing are not equivalent, as they each result in specific tumor phenotypes. The epigenetic inactivation of tumour suppressor genes by DNA hypermethylation seems to be tumour-type specific and affects all cellular pathways. One of the most compelling examples of the role of epigenetic gene silencing in the development of human cancer is the inactivation of DNA repair genes by promoter CpG island hypermethylation. As is true for mutations, gene regulation by epigenetic methylation can occur sporadically or it can be heritable.
Cancers usually arise from an assemblage of mutations and epimutations that confer a selective advantage leading to clonal expansion. Mutations, however, may not be as frequent in cancers as epigenetic alterations.
_
-28. Cellular systems to prevent cancer and their failure:
In addition to the controls on proliferation afforded by the coordinated action of proto-oncogenes and tumor suppressor genes, cells also have at least three other systems that can help them avoid runaway cell division.
The first of these systems is the DNA repair system.
A second cellular back-up system prompts a cell to commit suicide (undergo apoptosis) if some essential component is damaged or its control system is deregulated.
A third back-up system limits the number of times a cell can divide, and so assures that cells cannot reproduce endlessly. This system is governed by a counting mechanism that involves the DNA segments at the ends of chromosomes. Called telomeres, these segments shorten each time a chromosome replicates. The genetic program limits the number of times a cell is able to replicate, the so-called Hayflick limit, and when this limit is reached, replicative senescence is induced.
Cancer, then, does not develop all at once as a massive shift in cellular functions that results from a mutation in one or two wayward genes. Instead, it develops step-by-step, across time, as an accumulation of many molecular changes, each contributing some of the characteristics that eventually produce the malignant state.
A central feature of today’s molecular view of cancer is that cancer does not develop all at once, but across time, as a long and complex succession of genetic changes. Each change enables precancerous cells to acquire some of the traits that together create the malignant growth of cancer cells.
Mutations and epimutations in DNA repair genes allow DNA damage to cause mutations in regulatory genes. Mutations and epimutations in apoptotic genes prevent apoptosis. Genetic polymorphisms of cellular antioxidants are known to play a significant role in the pathogenesis of cancer.
Mutated proto-oncogenes become oncogenes, genes that stimulate excessive division. And mutations in tumor suppressor genes inactivate these genes, eliminating the critical inhibition of cell division that normally prevents excessive growth.
In general, human cells can go through only about 40-60 rounds of division before they lose the capacity to divide, “grow old,” and eventually die. Cancer cells can divide many more times than this, largely because they express an enzyme called telomerase, which reverses the wearing down of chromosome ends that normally happens during each cell division. In this context, immortality refers to the fact that, unlike normal cells which divide a finite number of times, cancer cells keep dividing more-or-less indefinitely. Telomerase mutations remove additional barriers, extending the number of times a cell can divide and generate a clonal cell population with a distinct advantage in proliferation.
Collectively, mutations in various genes account for much of the uncontrolled cell division that occurs in human cancers. The number of cell divisions that occur during this process can be astronomically large—human tumors often become apparent only after they have grown to a size of 10 billion to 100 billion cells. As you might expect, the time frame involved also is very long— it normally takes decades to accumulate enough mutations to reach a malignant state.
All in all, this paints a complex picture of how cancer starts. There are many things that can give a cell the potential to become cancerous, but there are checks in place to stop that from happening. Thus, many different regulatory systems have to be disrupted before a cell can throw off its normal restraints and behave defiantly as a malignant cancer cell. In addition, tumor cells may meet new barriers to further expansion at each stage of the evolutionary process. For example, oxygen and nutrients do not become limiting until a tumor is one or two millimeters in diameter, at which point the cells in the tumor interior may not have adequate access to such necessary resources. Each new barrier, whether physical or physiological, must be overcome by the acquisition of additional mutations. That is why so many mutations are needed to develop cancer.
_
-29. The immune system interacts closely with tumor cells over entire stages of cancer progression from tumor initiation and development to metastasis, facilitating either cancer inhibition or promotion. Immune cells and mediators of innate and adaptive immunity are essential components of the tumour microenvironment.
Medical research suggests that a strong immune system does protect against cancer. If we didn’t have an immune system, we’d all get cancer.
If the immune system is so strong and sophisticated, why does it fail so often in fighting cancer?
The short answer: because the cancer overpowers it.
Cancer cells can evade the host immune response by different mechanisms, among them are the production of immunosuppressive factors, low expression of tumor antigens, downregulation or aberrant expression of MHC class I on surface protecting it from cytotoxic effector functions of immune cells in the innate and adaptive immunity and co-stimulatory molecules. Cancer cells exert tremendous sway over some innate and adaptive immune cells and recruit them to help cancer grow and travel. There is a very intricate balance in the immune system that is usually anti-tumorigenic, meaning it eliminates tumors, but in some cases, if this balance is altered, these cells may actually help tumors grow and develop into full-blown metastatic disease. Innate and adaptive immunity in the tumour microenvironment are double-edged swords. Appropriately activated adaptive immune responses mediate resistance to carcinogenesis and progression. In contrast, cancer-related inflammation orchestrated by innate immunity, such as macrophages and the complement system, facilitates tumour progression via several mechanisms.
There is evidence to suggest that both activated oncogenes and mutated tumor suppressor genes actually trigger mechanisms that prevent immune system from rooting out cancer cells.
_
-30. Cancer treatment routinely involves taking out lymph nodes near the tumor in case they contain metastatic cancer cells. Lymph nodes are often removed because they are typically the first-place where metastatic cancer cells appear, and without surgery, it can be difficult to determine whether the nodes contain metastases. Most immunotherapies are aimed only at reinvigorating T cells in the tumor, where they often become exhausted battling the tumor’s cancer cells. Remember, T cells eliminate and edit cancer cells. T-cell cancer therapies already exist and the development of cancer immunotherapy has been one of the most exciting advances in the field. But the new research shows that allowing the immunotherapy treatment to activate the immune response of the lymph nodes as well can play an important role in driving positive response to immunotherapy. This research really changes our thinking about the importance of keeping lymph nodes in the body during treatment.
_
-31. The metastatic process accounts for the vast majority of deaths from solid tumors, and therefore, an understanding of this process is critical for improvements in survival from cancer. Cells that lose contact with the extracellular matrix (ECM) normally undergo programmed cell death (apoptosis induced by the loss of contact), and this process has to be suppressed in cells that metastasize. Another process is epithelial-mesenchymal transition (EMT). This is a process by which cells lose their epithelial properties and gain mesenchymal properties. Metastasizing epithelial cancer cells often undergo EMT as an important step in that process to become a cancer stem cell (CSC) capable of metastasis. CSC hypothesis posits that only CSC can initiate tumor formation and their daughter cells are incapable of forming new tumors by metastasis.
_
-32. Cancer cells exhibit poor use of oxygen and massive use of glucose. Malignant cells must focus a significant fraction of their energy resources into synthesis of proteins and other molecules (building blocks required for the production of new cells) while still maintaining sufficient ATP production to survive and grow. Although normal proliferating cells also have similar needs, there are differences in how cancer cells metabolize glucose and a number of other compounds including the amino acid glutamine as compared to normal cells in part because of genetic and epigenetic changes within cancer cells but also likely due to differences in the environments of cancer and normal cells. Normal cells typically generate only about 30% of energy from glycolysis, whereas most cancers rely on glycolysis for energy production. Many cancer cells utilize aerobic glycolysis (Warburg effect) to metabolize glucose, leading to increased lactic acid production, whereas normal cells utilize oxidative phosphorylation in mitochondria under aerobic conditions, a much more efficient process for generating ATP for energy utilization. Research shows that cancer cells consume glucose and amino acid glutamine at very high rates but these two molecules contribute little to the mass of new cells, and amino acids (excluding glutamine) contribute to the majority of the carbon atoms found in new cancer cells. It remains something of a mystery why proliferating human cancer cells consume so much glucose. If we can selectively block aerobic glycolysis in cancer cells, miraculous cure of most cancers can be found.
_
-33. Given that every cell division carries a risk of generating mutations, organisms with large bodies (composed of more cells) and extended longevities (with a longer time to accumulate mutations) should be more likely to develop cancer. Theory has it that the more the cells in an organism and the longer an organism lives, the more the likelihood of one of its cells succumbing to a random mutation and becoming cancerous. It would, therefore, be expected that larger organisms are more prone to cancers. Indeed, within humans and dogs, larger individuals are more likely to develop cancer than smaller ones. Similarly, increasing age is one of the most potent carcinogenic factors in species in which cancer aetiology is well studied. While elevated cancer risk with larger body size and/or longevity has been documented within species, Peto’s paradox indicates the apparent lack of such an association among taxa. Peto’s paradox is demonstrated by animals such as the bowhead whale, elephant, and the naked rat mole due to evolution of natural anticancer mechanisms.
_
-34. Cancer is prevalent in the animal kingdom. Cancer affects mammals, reptiles, birds, fish, and mollusks. On the other hand, several species are known to be extremely cancer resistant. These include the naked mole rat, blind mole rat, elephant, bats and bowhead whale. The known mechanisms of cancer resistance include duplications of the TP53 gene in elephants, overproduction of high molecular mass hyaluronan (HMM-HA) in the naked mole rat, interferon-mediated concerted cell death in the blind mole rat, and reduced growth hormone (GH)–insulin-like growth factor 1 (IGF1) signaling and microRNA (miRNA) changes in bats. Bowhead whale genome encodes a species-specific retroduplicated CDKN2CRTG gene that influence cellular phenotypes such as cell cycle progression and DNA damage repair in ways that are beneficial for aging and cancer resistance. Bowhead whales can live at least 211 years.
_
-35. Cancer cluster is defined as a greater-than-expected number of cancer cases that occurs within a group of people in a defined geographic area over a specific period of time. In a scientific review of over 500 cancer cluster investigations done over 20 years, only about 1 in 8 found a true increase in cancer rates, and in only one case was a clear cause for the increase found.
_
-36. When a person who already had cancer develops a new cancer, it is called a second cancer or second primary cancer. It is a completely new and different type of cancer than the first one. A second cancer is not the same as a cancer recurrence. A recurrence means that the first cancer has come back, in the same area of the body or in a different area. Reports and analyses of data have emphasized the importance of ruling out metastatic disease when a new tumor arises. One can readily appreciate a “second cancer” as such when the second neoplasm differs histologically, or molecularly, from the primary neoplasm. Second cancers have been known to result from the radiation and chemotherapy used to treat a primary cancer although most second cancers are due to other factors. Second cancers have become an increasingly important concern in oncology as they now comprise the sixth most common group of malignancies after skin, colorectal, lung, breast, and prostate cancers.
_
-37. Angiogenesis plays a critical role in the growth of cancer because solid tumors need a blood supply if they are to grow beyond a few millimeters in size. As tumor cells undergo dysregulated proliferation, the tumor mass initially expands beyond the support capacity of the existing vasculature, leading to decreased levels of oxygen and nutrients and accumulation of metabolic wastes. Tumor cells respond to this deterioration of the tumor microenvironment by up-regulating several proangiogenic factors, including vascular endothelial growth factor, basic fibroblast growth factor, placental growth factor, and platelet-derived endothelial growth factor, which collectively activate quiescent endothelial cells and promote their migration into the tumor. This shift of the tumor microenvironment to an angiogenic state, or ‘‘angiogenic switch’’, is an important rate limiting factor in tumor development. Tumor metastasis is also regulated by angiogenesis, as well as by lymphangiogenesis, where new lymphatic vessels are formed from preexisting ones. If we can selectively block angiogenesis in solid tumors, we can find miraculous cure of most solid cancers.
_
-38. Whether or not inflammation is a cause or a consequence of cancer, tumor microenvironment (TME) triggers an immune inflammatory response, and histopathological analyses provide evidence for the presence of innate and adaptive immune cells in most human tumors.
The triggers of chronic inflammation that increase cancer risk or progression include infections (e.g., Helicobacter pylori for gastric cancer and mucosal lymphoma; papilloma virus and hepatitis viruses for cervical and liver carcinoma, respectively), autoimmune diseases (e.g., inflammatory bowel disease for colon cancer) and inflammatory conditions of uncertain origin (e.g., prostatitis for prostate cancer). NSAIDs lower cancer risk by multiple mechanisms. NSAIDs reduce the incidence of colonic adenomas and colorectal cancer (CRC).
Inflammation contributes to tumor growth and survival by supplying factors that sustain proliferation; factors that limit cell death; proangiogenic factors; extracellular matrix-modifying enzymes that facilitate angiogenesis, invasion, and metastasis; and other signals that lead to activation of EMT and other hallmark-facilitating programs. Inflammatory cells also release notably reactive oxygen species that are actively mutagenic for nearby cancer cells, accelerating their genetic evolution toward states of heightened malignancy. An additional mechanism involved in cancer-related inflammation (CRI) is induction of genetic instability by inflammatory mediators, leading to accumulation of random genetic alterations in cancer cells.
Chronic inflammation is a necessary consequence of cancer progression. Chronic inflammation is a critical hallmark of cancer, with at least 25% of cancers associated with it.
_
-39. Age is a prominent risk factor for most types of cancer, including breast, lung and colon cancers, which each have a large impact on public health. Cancer risk increases with age, in part, because genetic mutations that arise from DNA replication errors and exposure to environmental carcinogens accumulate as we get older. Additionally, less effective DNA repair mechanisms come with aging and progressive decay of immune function occurs in older individuals, whereby an effective immune response against developing tumors may fail.
_
-40. It is now clear that aging and cancer development either share or diverge in several disease mechanisms. Such mechanisms include genomic instability, telomere attrition, epigenetic changes, loss of proteostasis, decreased nutrient sensing, altered metabolism, cellular senescence and stem cell function. Cancer cells and aged cells are also fundamentally opposite, as cancer cells can be thought of as hyperactive cells with advantageous mutations, rapid cell division and increased energy consumption, while aged cells are hypoactive with accumulated disadvantageous mutations, cell division inability and a decreased ability for energy production and consumption. The underlying mechanism in both cancer and aging is the time dependent accumulation of cellular damage. Several hallmarks of aging (i.e., genomic instability, epigenetic alterations, chronic inflammation, and dysbiosis) are very similar to specific cancer hallmarks and hence constitute common “meta-hallmarks,” while other features of aging (i.e., telomere attrition and stem cell exhaustion) act likely to suppress oncogenesis and hence can be viewed as preponderantly “antagonistic hallmarks.” The risk of developing cancer appears to increase until the age of 70 to 80 and then diminish. This is explained by the agonistic and antagonistic relationship between the processes that drive aging and cancer.
_
-41. Only 5–10% of all cancers are due to an inherited gene defect. The majority of cancers, some 90–95% of cases, are due to genetic mutations from environmental factors. The concordance between identical twins for breast cancer was found to be only 20%. Common environmental factors that contribute to cancer death include tobacco use (25–30%), diet and obesity (30–35%), infections (15–20%), radiation (both ionizing and non-ionizing, up to 10%), lack of physical activity, and pollution.
Various twin studies found that overall, about 30% of cancers are familial and 70% are sporadic. But there is a difference between familial cancers and inherited cancers. Familial cancer is used to describe a situation in which more members of a particular family are diagnosed with a type of cancer than would be statistically expected; hereditary and lifestyle causes separately or together may contribute to the high incidence in the family. In contrast, the term inherited cancer is used to refer to familial cancers in which a genetic cause has been identified. In other words, out of 30 % cancers that are familial in twin studies, not all are due inherited genetic defect and many are due to common lifestyle factors as twins lived in the same family.
_
-42. It is not generally possible to prove what caused a particular cancer because the various causes do not have specific fingerprints. For example, if a person who uses tobacco heavily develops lung cancer, then it was probably caused by the tobacco use, but since everyone has a small chance of developing lung cancer as a result of air pollution or radiation, the cancer may have developed for one of those reasons.
_
-43. Carcinogens are agents in the environment capable of contributing to cancer growth. These fall into several broad groups, which can be categorized as infective (e.g., viruses), chemical (including occupational, environmental, and therapeutic), electromagnetic radiation (i.e., ultraviolet [UV], X-ray, gamma ray), and immunosuppressive (HIV, immunosuppressive medications). Several hundred chemicals have been shown to be carcinogenic in humans. Harm from these chemical carcinogens can be a result of occupational (asbestos, aniline dyes), environmental (alcohol, tobacco), or iatrogenic (chemotherapy) exposure. While many carcinogens are directly mutagenic to DNA, most carcinogens undergo activation after exposure to reactive metabolites that are responsible for genetic damage.
_
-44. Approximately 20% of all cancers are associated with infectious agents, and 12% of all cancers are caused by oncoviruses. Oncovirus infections are common, but these infections rarely result in cancer. One or more additional insults, such as chronic inflammation, environmental mutagens, or immunosuppression, are required for cancer development. Some cancers often require cofactors as well as the virus to develop. For example, in the case of HPV, cofactors in the development of cervical cancer include smoking, hormonal contraceptives, nutrition, and coinfections with other organisms, such as herpesvirus, Chlamydia, and HIV.
Viral cancers do not arise acutely after infection, but instead develop 15–40 years later. One exception is a rare EBV-associated lymphoproliferative disease, which can occur shortly after infection. In cancers, viral replication is either diminished or absent, as active replication would lyse the host cell and prevent tumorigenesis. The virus exists intracellularly as naked nucleic acid in the form of a plasmid, episome, or cellular-integrated genome.
In general, all oncoviruses promote tumorigenesis via common pathways. Tumor suppressor pathways, such as p53 and retinoblastoma 1(Rb1), are often inhibited.
Viruses have adapted by evolving different strategies for evading immune responses. In general, it can be said that these mechanisms are also involved in the immune evasion of virally induced cancers and that this is not because the virus evolved to cause cancer but rather is a secondary result of the mechanisms that the virus deployed to evade the elimination of virally infected cells.
Certain bacterial infections also increase the risk of cancer, as seen in Helicobacter pylori-induced gastric carcinoma. The mechanism by which H. pylori causes cancer may involve chronic inflammation or the direct action of some of the bacteria’s virulence factors. Parasitic infections strongly associated with cancer include Schistosoma haematobium (squamous cell carcinoma of the bladder) and the liver flukes, Opisthorchis viverrini and Clonorchis sinensis (cholangiocarcinoma).
There is an increased risk of certain types of non-infectious cancers in HIV-1-infected individuals, presumably due to the defective cell-mediated immunity, but HIV-1 is not considered an oncovirus.
_
-45. Free radical is atom or group of atoms that has at least one unpaired electron and is therefore unstable and highly reactive. Free radicals are formed naturally in the body and play an important role in many normal cellular processes. At high concentrations, however, free radicals can be hazardous to the body and damage all major components of cells, including DNA, proteins, and cell membranes. The damage to cells caused by free radicals, especially the damage to DNA, may play a role in the development of cancer and other health conditions. Free radicals that contain the element oxygen are the most common type of free radicals produced in living tissue. Another name for them is “reactive oxygen species,” or “ROS”. Oxidative stress can lead to mutations in DNA, and more than 100 different types of oxidative damage to DNA have been identified. The induction of oxidative stress and subsequent injury is a characteristic of a diverse group of carcinogens, including radiation, asbestos, chemicals, and carcinogenic infectious agents.
_
-46. Poverty is a great risk factor in Human Cancers. Poor people’s low socioeconomic status typically means a low intake of garden-fresh fruit and vegetables which is associated with a higher risk of gastro intestinal cancers. In addition, poor people are more vulnerable to environmental factors inducing human cancers such as tobacco use, sunlight and ionizing radiation, alcohol consumption, organic and inorganic chemicals, and infectious micro-organisms. On the other hand, people with extreme poverty and calorie restriction have lower risk of cancer (vide infra) but they may succumb to malnutrition and infections.
_
-47. Tobacco use is the leading preventable cause of cancer and cancer deaths. It can cause not only lung cancer — but also cancers of the mouth and throat, voice box, esophagus, stomach, kidney, pancreas, liver, bladder, cervix, colon and rectum, and a type of leukemia. Carcinogens from tobacco products can be taken in directly through inhalation or ingestion (smokeless tobacco) and also may be absorbed into the circulation. Metabolites formed during the activation of carcinogens may bind covalently with DNA to form DNA adducts, usually at adenine or guanine. If DNA adducts escape cellular repair mechanisms they could persist and may lead to miscoding, resulting in a mutation. In addition, cigarette smoke contains free radicals that can induce oxidative damage of DNA in humans and cause mutations. Certain mutations could trigger the activation of an oncogene or the deactivation of a tumour suppressor gene. Tobacco smoking may result in impairment of the immune system functioning thereby increasing the risk of cancer.
Lung cancer death rates in the United States have mirrored smoking patterns, with increases in smoking followed by dramatic increases in lung cancer death rates and, more recently, decreases in smoking rates since the 1950s followed by decreases in lung cancer death rates in men since 1990.
The fact that not all tobacco-users develop cancer is frequently used to argue against a role of tobacco in carcinogenesis. The real reason for this (aside from the random element) is that characteristics relating to individual smokers may put them at increased risk of cancer, or indeed, may be protective, and these are called cofactors. These cofactors include age, diet, occupation, environment and genetic cofactors.
_
-48. Alcohol is a carcinogen and increases cancer incidence and mortality. In Western Europe 10% of cancers in males and 3% of cancers in females are attributed to alcohol. Worldwide, 3.6% of all cancer cases and 3.5% of cancer deaths are attributable to alcohol. In particular, alcohol use has been shown to increase the risk of developing cancers of the mouth, esophagus, pharynx, larynx, stomach, liver, ovaries, and colon. Alcohol consumption also significantly increases the risk for breast cancer. The association is stronger for the hormone-sensitive form of breast cancer than for the insensitive form. The main mechanism of cancer development involves increased exposure to acetaldehyde, a carcinogen and breakdown product of ethanol. Other mechanisms have been proposed, including alcohol-related nutritional deficiencies, changes in DNA methylation, induction of oxidative stress in tissues and interfering with oestrogen metabolism.
_
-49. Overweight and obesity accounted for 14 percent of all cancer deaths in men and 20 percent of those in women. Significant positive associations were found between obesity and higher death rates for the following cancers: esophagus, colon and rectum, liver, gallbladder, pancreas, kidney, stomach (in men), prostate, breast, uterus, cervix, and ovary. Obesity leads to a limited but real increase of cancer risk and mortality, possibly due to over-stimulation of mTOR-related pathways. Calorie restriction reduces the exposure of cells to high glucose levels, insulin and other growth factors, favouring a metabolic state dominated by AMPK, while at the same time inhibiting mTOR activity. It is no surprise, therefore, that calorie reduction does lower the frequency of spontaneous and induced tumours in laboratory animals. For obvious reasons, there is only circumstantial evidence that drastic calorie reduction has the same effect in humans. The example most often cited is a study of the population of Okinawa with a total energy intake 20% below the Japanese (and 40% below the US) average. They live longer, and death rates due to cancer are 30% lower than in the rest of Japan. Studies have found that bariatric surgery among people with obesity, particularly women, is associated with reduced risks of cancer overall; of hormone-related cancers, such as breast, endometrial, and prostate cancers; and of obesity-related cancers, such as postmenopausal breast cancer, endometrial cancer, and colon cancer.
_
-50. A high fruit and vegetable diet would reduce cancers of the mouth and pharynx, esophagus, lung, stomach, and colon and rectum; evidence of probable risk reduction was found for cancers of the larynx, pancreas, breast, and bladder. The inverse correlation between the intake of fruits and vegetables, and the consumption of alcohol and tobacco is a potent confounding factor which cannot be overlooked.
_
-51. Establishing a causal relationship between a particular diet and cancer is difficult. Cancer is a chronic disease surfacing after many years of (possibly) harmful eating behaviour. This practically excludes the kind of controlled experiments demanded in science and leaves us with mainly observational studies that are flawed.
_
-52. Studies have linked eating red or processed meat to an increased risk of breast cancer, colon cancer, prostate cancer, and pancreatic cancer. Not coincidentally, carnivorous animals have the highest cancer-related mortality.
_
-53. There is currently no compelling evidence that supplementing vitamins, antioxidants or other micronutrients reduces cancer incidence with possible exceptions of vitamin D and folates.
_
-54. Tea polyphenols have been shown to inhibit tumor cell proliferation and induce apoptosis in laboratory and animal studies. In other laboratory and animal studies, tea catechins have been shown to inhibit angiogenesis and tumor cell invasiveness. More than 50 epidemiologic studies of the association between tea consumption and cancer risk have been published since 2006. The results of these studies have often been inconsistent, but some have linked tea consumption to reduced risks of cancers of the colon, breast, ovary, prostate, and lung. Coffee consumption has been associated with reduced risk for hepatocellular cancer.
_
-55. There is strong evidence that higher levels of physical activity are linked to lower risk of several types of cancer. Patients who exercise regularly have a 21 – 35% lower relative risk of cancer recurrence, a 28 – 44% reduced relative risk of cancer-specific mortality and a 25 – 48% decreased relative risk of all-cause mortality.
_
-56. Substantial and convincing bodies of experimental, clinical, and epidemiologic evidence indicate that hormones play a major role in the etiology of several human cancers. Evidence is mounting to show that the amount of hormone to which a tissue is effectively exposed is under strong genetic control. Therefore, in addition to external factors such as diet or exogenous hormone use which may modify hormone profiles, polymorphisms in genes encoding proteins involved in steroid-hormone biosynthesis, metabolism or extra- and intracellular transport, and DNA binding are important determinants of individual cancer risk. Hormones are important agents in sex-related cancers such as cancer of the breast, endometrium, prostate, ovary, and testis, and also of thyroid cancer and bone cancer. Women who take hormone replacement therapy have a higher risk of developing cancers associated with those hormones.
_
-57. The carcinogenic activity of UV light from solar radiation is attributable to the formation of pyrimidine dimers in DNA. If a pyrimidine dimer in a growth regulatory gene is not immediately repaired, it can contribute to tumour development. Fair-skinned persons exposed to the sun have the highest incidence of melanoma because they have the least amount of protective melanin.
_
-58. Besides direct DNA damage, various molecules broken by ionizing radiation can become highly reactive free radicals that cause further chemical damage. Some of this direct and indirect damage will eventually impact chromosomes and epigenetic factors that control the expression of genes. Cellular mechanisms will repair some of this damage, but some repairs will be incorrect and some chromosome abnormalities will turn out to be irreversible. DNA double-strand breaks (DSBs) are generally accepted to be the most biologically significant lesion by which ionizing radiation causes cancer. Research suggests that mutagenic events do not occur immediately after irradiation. Instead, surviving cells appear to have acquired a genomic instability which causes an increased rate of mutations in future generations. The cell will then progress through multiple stages of neoplastic transformation that may culminate into a tumor after years of incubation. Cancers associated with high dose ionizing radiation exposure include leukemia, breast, bladder, colon, liver, lung, esophagus, ovarian, multiple myeloma, and stomach cancers.
_
-59. Our everyday ordinary risk of cancer is about 42 percent without radiation exposure over a lifetime. This increases to 59% after 1 sievert (1000mSv) ionizing radiation exposure. That is 17% increase per sievert according to International Commission on Radiological Protection (ICRP).
However, the consensus of the nuclear industry, nuclear regulators, and governments, is that the incidence of cancers due to ionizing radiation can be modelled as increasing linearly with effective radiation dose at a rate of 5.5% per sievert.
_
-60. CT imaging involves the use of x-rays, which are a form of ionizing radiation. Exposure to ionizing radiation is known to increase the risk of cancer. The effective doses from diagnostic CT procedures are typically estimated to be in the range of 1 to 10 mSv. Going by ICRP calculations, 10 mSv CT exposure increases cancer risk by 0.17%. For comparison, a single chest x-ray exposes the patient to about 0.1 mSv while a regular-dose CT scan of the chest is 7 mSv, low-dose CT chest is 1.5 to 2 mSv, and effective radiation dose of ultra-low-dose chest CT can be as low as 0.14–0.5 mSv. Ionizing radiation exposure from coronary CT angiography is 16 mSv.
______
Dr Rajiv Desai.MD.
May 21, 2023
_____
Postscript:
Cancer is a genetic disorder caused predominantly by environmental factors. Studies with identical twins have suggested that genes are not the source of most chronic illnesses. Cell functions are not exclusively genetically determined, but are a consequence of molecular interactions, reactions, and biophysical influences that act on the molecules in the immediate environment. Most biology come from the complex interaction of all the proteins and cells working with environmental factors, not driven directly by the genetic code. All those who persecuted me on the basis of my DNA have made a big mistake. My character is not exclusively determined by my DNA, but by interaction of DNA with environment which includes family, friends, neighbors, teachers, community, hometown, religion and culture.
_____
Footnote:
This article discusses the processes that lead to cancer and the more we learn about these processes, the more opportunities arise to intervene and attack cancer in ever more sophisticated and precise ways. That’s why understanding the basic biology of cancer is so fundamental to making progress against it. Whether or not there will ever be a cure for all cancer types is currently a matter of strong debate; but as cancer types vary immensely, it makes it very difficult to say that an approach that works for one type will be adaptable to all. Also, while there is much emerging research promising more effective treatments, most of these projects are still in their early stages, having conducted in vitro and in vivo experiments. Some potential treatments still have a long way to go before clinical trials in human patients. Still, that doesn’t mean we should lose all hope. Some researchers explain that these efforts should make us optimistic; while we may not be at the stage where we can claim that cancer can easily be eradicated, our furthered knowledge and ever more precise tools keep us ahead of the game and improve our odds in the fight against this disease.
_____
_____
3 comments on “What Causes Cancer”
Leave a Reply

как пишется никак слитно или раздельно правильно
http://www.openkrokzo.ru
высота в треугольнике как найти
[url=http://www.mirtetriks.ru]mirtetriks.ru[/url]
http://openkrokzo.ru
о чем книга белые ночи достоевского
[url=https://frutilupik.ru]frutilupik.ru[/url]
http://sigmablog.ru
где находится 3 4 пресной воды в гидросфере
[url=http://webgraal.ru]toktiblog.ru[/url]
http://www.fokusblog.ru
что называется линиями магнитной индукции их свойства
Remarkable! Itts really remarkable paragraph, I
hve got much clear idea about from this post.
Amazing! This blog looks just like my old one! It’s on a
totally different topic but it has pretty much the same page
layout and design. Great choice of colors!