Dr Rajiv Desai
An Educational Blog
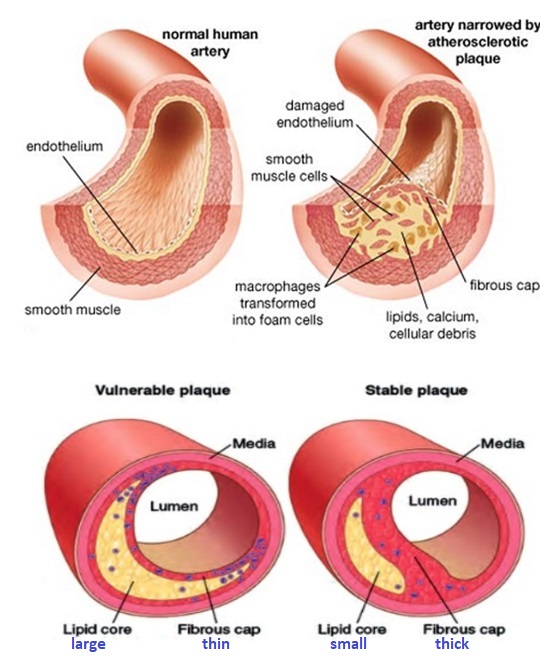
VULNERABLE PLAQUE
Vulnerable Plaque:
____
____
Prologue:
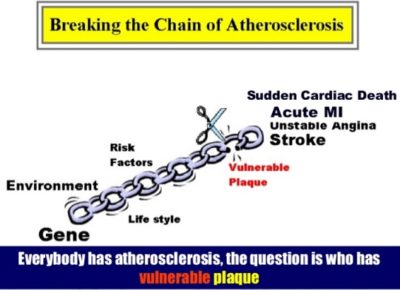
Worldwide more than 17 million people die every year from cardiovascular disease (CVD) with this number projected to increase to over 23 million by 2030. The vast majority of acute ischemic vascular events occur in relation to an underlying atherosclerotic plaque. Atherosclerotic plaque represents the hallmark lesion of atherosclerosis. Plaque rupture is the dominant initiating event, responsible for 60–70% of acute coronary syndromes (ACS), while plaque erosion is responsible for most of the remainder. Irrespective of the mechanism, the consequence is exposure of a thrombogenic substrate to circulating blood. This in turn triggers platelet aggregation and the coagulation cascade which compromises vascular blood flow resulting in downstream end-organ ischemia and infarction. These events occur abruptly and often without warning. Despite major advances in treatment of coronary artery disease patients, a large number of victims of the disease who are apparently healthy die suddenly without prior symptoms. Available screening and diagnostic methods are insufficient to identify the victims before the event occurs. The patient’s risk factors (e.g. smoking, high lipids, diabetes and hypertension) are very important to plaque stability, in addition to the plaque components. However, traditional risk factor-based screening fails miserably to predict major adverse cardiovascular events (MACE). Take a group of patients who arrive at the ER with no prior MI, CAD, diabetes, or other symptoms. Turn back the clock 24 hours, and run them through the existing guidelines for risk stratification, and only about 12% fall into the high-risk category and about half would not even qualify for treatment. Approximately 70% of people would have been categorized as low risk. Take example of cigar-smoking, overweight Winston Churchill and the runner Jim Fixx, who was very fit. Churchill did not suffer a major event until age 90, while Fixx died of a massive MI at age 53.
Until recently, the high-risk plaque has only been identified by retrospective analysis, predominantly from pathological examination of autopsy specimens. This has limited our ability to appreciate the dynamic nature of plaque vulnerability and rupture, and has placed a heavy reliance on invasive angiography to describe the anatomical luminal stenosis severity rather than plaque biology. To date, cardiologists have alleviated the symptoms of patients with significant lesions in the coronary angiography by stenting or coronary artery bypass grafting. While normal-looking coronary segments in angiography have been regarded as ‘disease-free’ and mild/moderate stenoses as ‘non-treatable’, today we know that from these non-significantly diseased areas may potentially arise acute coronary events if no further action is taken other than standard care for cardiovascular risk factors. Indeed, despite intensive management, many patients continue to experience recurrent coronary events.
The term “vulnerable plaque” refers to asymptomatic atherosclerotic plaque that is prone to rupture/erode and may result in life-threatening events which include myocardial infarction and sudden cardiac death. Vulnerable plaques are commonly found in coronary arteries at autopsy but are virtually undetectable by standard diagnostic techniques such as stress testing and coronary angiography.
Early identification and treatment of the vulnerable plaque has gained great interest in the cardiovascular research field. Recent advances in invasive and noninvasive coronary imaging techniques have empowered the clinician to identify suspected vulnerable plaques in vivo and paved the way for the evaluation of therapeutic agents targeted at reducing plaque vulnerability. The treatment of acute coronary syndromes, including acute myocardial infarction and unstable angina, has been one of the most successful contributions in the last 20 years to the improvement of medicine. However, we are still unable to predict them, and therefore to prevent occurrence of acute myocardial infarction and/or death. The concept of vulnerable plaque has helped us to understand better the cascade resulting in acute occlusion.
______
Note:
In this article, discussion about vulnerable plaque would revolve around vulnerable coronary plaque.
______
______
Abbreviations and synonym:
VP = vulnerable plaque
AMI = acute myocardial infarction
STEMI = ST segment elevation myocardial infarction
NSTEMI = non-ST segment elevation myocardial infarction
CAD = coronary artery disease
CVD = cardiovascular disease
ACS = acute coronary syndrome
ER = emergency room
US = ultrasound
ACE = angiotensin converting enzyme
CCTA= coronary computed tomography angiography = CTCA = CT coronary angiography
EBCT= electron-beam computed tomography
FFR= fractional flow reserve
hsCRP = high-sensitive C-Reactive Protein
IL= interleukin
IVUS = intravascular ultrasound
VH = virtual histology
M-CSF= Macrophage Colony-Stimulating Factor
Hu = Hounsfield units
RI = remodeling index
LAP = low attenuation plaque
FDG = flurodeoxyglucose
MRI= Magnetic Resonance Imaging
MSCT= Multi-Slice Computed Tomography = MDCT = multidetector computed tomography
OCT= Optical Coherence Tomography
PET= Positron-Emission Tomography
PET-CT= Positron-Emission Tomography Computed Tomography
SPECT = Single positron emission computed tomography
MR = Microwave radiometry
NIRS = Near infrared spectroscopy.
HDL = high-density lipoprotein
LDL = low-density lipoprotein
MCP-1= monocyte chemotactic protein-1
NOS = nitric oxide synthase
VCAM-1 = vascular cell adhesion molecule-1
CL = collagenase
GL = gelatinase
ECM = extracellular matrix
MMP = matrix metalloproteinase
SMC = smooth muscle cell
TCFA = thin cap fibroatheroma
IEL = internal elastic lamila
IPH = intraplaque hemorrhage
CC = cholesterol crystals
CAC = coronary artery calcium
ESS = endothelial (wall) shear stress
Circumferential wall tension = wall tension = blood pressure X radius of artery
AHA = American heart association
PCI = percutaneous coronary intervention
MACE = Major adverse cardiovascular events, a criterion for evaluating cardiovascular disease treatments such as angioplasty.
Centimeter square = cm2 = cm2
_______
_______
Atherosclerosis:
Atherosclerosis is a systemic disease of the vessel wall. The main components of the atherosclerotic plaque are connective tissue extracellular matrix, including collagen, proteoglycans, and fibronectin elastic fibers; cholesterol, cholesteryl esters, and phospholipids; and cells such as monocyte-derived macrophages, T lymphocytes, and smooth muscle cells. Varying proportions of these components occur in different plaques, giving rise to a spectrum of lesions.

Arteriosclerosis is any hardening (and loss of elasticity) of medium or large arteries (from the Greek arteria, meaning artery, and sclerosis, meaning hardening). Atherosclerosis is a hardening of an artery specifically due to an atheromatous plaque. The term atherogenic is used for substances or processes that cause atherosclerosis. Atherosclerosis is a specific type of arteriosclerosis.
_
Structure of normal arteries:
In mammals, the arterial wall is constituted by three histological layers.
- The intima (or tunica intima), the innermost layer in contact with the blood flow, is constituted by a monolayer of endothelial cells and a subendothelial connective tissue layer limited by the internal elastic lamina. Endothelial cells are joined by tight junctions that participate in intercellular cohesion and by gap junctions involved in intercellular electrochemical coupling. In the normal arterial wall, the subendothelial ECM is a thin layer of connective tissue that constitutes an adhesive scaffold required for the anchorage-dependent survival of endothelial cells, a reservoir of growth factors and a transducer of physical and biochemical changes of the microenvironment. Intimal thickening is one of the earliest stages of atherosclerosis.
- The intermediate layer, tunica media, is mainly constituted by SMC and ECM components, including elastin, collagen and proteoglycans. It is separated from the tunica intima by the internal elastic lamina and from the adventitia by the external elastic lamina. In medium-sized muscular arteries, the media is mainly constituted by smooth muscle cells (SMC) surrounded by ECM and its thickness is correlated with the diameter of arteries. The media of the aorta consists of concentric musculoelastic layers that serve to the biomechanical properties (viscoelasticity) of the aortic wall.
- The adventitia (tunica adventitia) is the outer layer of the vessel. It is constituted by fibroblasts and a loose connective tissue that contains vasa vasorum. Vasa vasorum are derived from the same vessel or a neighboring vessel (artery or vein), run along the arterial wall and penetrate into the adventitia of arteries where they supply oxygen and nutrients to the vascular wall. In the thoracic aorta, these microvessels penetrate up to 2/3 of the external media, while the intima and the inner part of the media are nourished by diffusion from the vascular lumen. In intimal hyperplasia and atherosclerotic plaque, the vasa vasorum network is extended and penetrates the media and the pathological intima.
_
Atherogenesis is a slowly progressive process characterized by multifocal structural alterations of the vascular wall of medium and large arteries, leading to the formation of atherosclerotic plaques. The pathogenic events of atherogenesis associate endothelial dysfunction and activation, monocyte/macrophage adhesion, activation and migration, local oxidative stress, lipid deposition, extracellular matrix (ECM) synthesis, smooth muscle cell (SMC) migration and proliferation and neovascularization of the plaque. Atherosclerotic plaque rupture is the primary mechanism responsible for two of the biggest killers worldwide: myocardial infarction and stroke. Whilst the clinical effects of atherosclerotic plaque rupture can be devastating, the development of atheromatous plaque is itself a silent and for many a benign process. Indeed, atherosclerosis is an almost ubiquitous finding in older patients the majority of whom will never suffer a cardiovascular event.
_
The working definition of atherosclerosis:
Atherosclerosis is a systemic dysfunctional endothelial, focal occurring, chronic inflammatory, fibro-proliferative, prothrombotic, angiogenic, multifactorial disease of the arterial intima caused by the retention of modified low-density lipoproteins, hemodynamic, and reductive-oxidative (redox) stress. There is no question that atherosclerosis is a systemic dysfunctional endothelial disease. It is focal, in that, lesions have a tendency to occur at predictable anatomic sites of the arterial tree. It predictably occurs at bifurcations, side branches, and opposite flow dividers at areas of low endothelial shear stress and turbulent blood flow. There is an orderly cephalad progression over time starting with the iliacs and progressing cephalad to the aorta, coronaries, carotids and cerebral vessels.
_
Atherosclerosis is a multifocal, smoldering, immunoinflammatory disease of medium-sized and large arteries fueled by lipids. Endothelial cells, leukocytes, and intimal smooth muscle cells are the major players in the development of this disease. The most devastating consequences of atherosclerosis, such as heart attack and stroke, are caused by superimposed thrombosis. Therefore, the vital question is not why atherosclerosis develops but rather why atherosclerosis, after years of indolent growth, suddenly becomes complicated with luminal thrombosis. If thrombosis-prone plaques could be detected and thrombosis averted, atherosclerosis would be a much more benign disease. Approximately 76% of all fatal coronary thrombi are precipitated by plaque rupture. Plaque rupture is a more frequent cause of coronary thrombosis in men (80%) than in women (60%). Ruptured plaques are characterized by a large lipid-rich core, a thin fibrous cap that contains few smooth muscle cells and many macrophages, angiogenesis, adventitial inflammation, and outward remodeling. Plaque rupture is the most common cause of coronary thrombosis. Ruptured plaques and, by inference, rupture-prone plaques have characteristic pathoanatomical features that might be useful for their detection in vivo by imaging.
Atherosclerosis is by far the most frequent underlying cause of coronary artery disease, carotid artery disease, and peripheral arterial disease. Atherosclerosis alone is rarely fatal; it is thrombosis, superimposed on a ruptured or eroded atherosclerotic plaque, that precipitates the life-threatening clinical events such as acute coronary syndromes and stroke. Therefore, the vital question is not why atherosclerosis develops but rather why none or only few among many plaques within a given person apparently pass through a thrombosis-prone and dangerous phase during a lifetime. That is what it is all about; preventing the development of thrombosis-prone plaques and, if they already are there, finding and treating those who harbor them and are at high risk of losing myocardium, brain, and/or life.
_
Atherosclerosis is a multifocal slowly progressive process affecting the intima of medium-sized and large arteries. This chronic metabolic and inflammatory process is characterized by the formation of plaques constituted by a cholesterol-rich core (atheroma) surrounded by a fibrous cap (sclerosis). The histological classification describes the progression of lesions: types I and II are early lesions (intimal thickening and fatty streaks), whereas types II to VI lesions correspond to advanced lesions (fibro-lipidic, calcified and complicated plaques). Atherosclerosis is a smouldering immunoinflammatory disease fuelled by lipids. It is characterised by focal thickening of the arterial intima (plaque formation) in medium and large sized arteries. Within the plaques lipid, inflammatory infiltrates, smooth muscle cells and connective tissues are found. An injury to the plaque cap known as a plaque rupture results in exposure of its core contents to the blood, causing acute thrombus formation and either partial or complete occlusion of the vessel lumen. Atherothrombosis from plaque rupture is the most common cause of fatal myocardial infarction, accounting for approximately three quarters of cases, with plaque erosion accounting for the remaining quarter. However, the majority of coronary plaque rupture events appear to be clinically silent, resulting in plaque growth rather than myocardial infarction.
_
The initial trigger of atherogenesis results apparently from the hemodynamic stress, i.e. turbulent blood flow, which elicits endothelial cell activation in atherosclerosis prone areas (arterial bifurcations). The activated endothelium exhibits an increased permeability, generates reactive oxygen species (ROS) and expresses inflammatory adhesion proteins and chemokines. The endothelium permeability allows an influx of plasma components, in the subendothelial area, where lipoproteins undergo various modifications, including oxidation. Chemokines and adhesion proteins promote the recruitment of leukocytes. Monocytes take up modified lipoproteins, accumulate lipids (mainly cholesterol esters) and are converted into macrophagic foam cells that form fatty streaks. These early lesions may rapidly grow in case of hypercholesterolemia, or may regress if the LDL-cholesterol and other pro-atherogenic factors decrease.
Atherosclerosis begins when low-density lipoproteins (LDL) infiltrate the endothelial wall. Subsequent oxidation of LDL molecules causes an inflammatory response with infiltration of T-lymphocytes and macrophages that consume LDL and form foam cells. This is initially protective, but with further LDL accumulation, macrophage cell death is ultimately triggered, contributing to further inflammation and the development of a necrotic core of soft unstable atheroma. Atherogenesis starts early in infancy, and evolves slowly over decades, leading progressively to the formation of plaques characterized by a lipid-rich core surrounded by a fibrous cap constituted by ECM proteins secreted by proliferating SMC in the intima and myofibroblasts. Cholesterol deposition, associated with a local inflammatory response and the secretion of pro-inflammatory cytokines, promotes the progression of atheromatous plaques. Moreover, neoangiogenesis may play a role in plaque growth and complication, as suggested by the study of neovascularization in atherosclerotic lesions and in ruptured plaques associated with thrombotic events. Plaque inflammation also triggers smooth muscle cell loss and the production of matrix metalloproteinases (MMP) that weaken the fibrous cap, predisposing it to plaque rupture. The thick necrotic acellular lipid core becomes increasingly hypoxic, stimulating angiogenesis, with the formation of immature microvessels prone to intra-plaque haemorrhage (IPH).
Similar to tuberculosis, calcification of atherosclerotic plaque is thought to be a healing response to intense necrotic plaque inflammation characterised by two distinct stages. In the latter stage of macrocalcification, the healing process is complete and the plaque stabilised. By contrast, the earlier stage of microcalcification is a common feature of ruptured and unstable plaques where healing is incomplete, inflammation remains active and the fibrous cap weakened by the tiny calcific deposits.
Unstable plaques at risk of rupture, therefore, have certain pathological features, including a large necrotic core, thin fibrous cap, inflammation, hypoxia, hemorrhage and microcalcification. By contrast, stable plaques at low risk of rupture have different characteristics, including a thick fibrous cap and macroscopic calcification.
_
Pathogenesis of atherosclerosis in a nutshell:
The disease is initiated by the activation of the endothelium/endothelial cell (EC) dysfunction by accumulation of LDL, which subsequently gets modified (e.g. oxidized), together with other atherogenic factors. The activated ECs secrete a range of chemokines and increase the expression of adhesion proteins on their cell surface. This results in the recruitment and infiltration of immune cells such as monocytes. The monocytes differentiate into macrophages, which is accompanied by increased expression of pattern recognition receptors on their surface, which participate in the promotion of inflammation and uptake of modified LDL, leading to the formation of lipid laden foam cells. Continued accumulation of modified LDL together with disturbed cellular lipid homeostasis causes apoptosis/necrosis of foam cells resulting in lipid deposition (necrotic core) and amplification of the inflammatory response. Smooth muscle cells (SMCs) migrate from the media to the intima where they proliferate, uptake modified lipoproteins and secrete extracellular matrix (ECM) proteins that stabilizes the plaques (fibrous cap). Continued inflammation orchestrated by cytokines destabilizes such plaques via decreased production of ECM proteins (reduced synthesis together with apoptosis/necrosis of SMCs/SMC-derived foam cells), increased production/activities of ECM degrading matrix metalloproteinases (MMPs) and reduced expression/activities of inhibitors of these enzymes. Plaque rupture leads to platelet aggregation, coagulation and thrombus formation that ultimately results in the clinical complications associated with this disease. Cytokines affect all the different stages in the pathogenesis of atherosclerosis.
_______
Atherosclerotic risk factors — are there ten, or is there only one? A 1992 study:
The Expert Panel of the National Cholesterol Education Program has identified 10 risk factors for the occurrence of an atherosclerotic event. Each of these factors does not represent an independent risk. Male sex, family history of premature coronary events, cigarette smoking (> 10/day), systemic hypertension, diabetes mellitus and severe obesity (>30% overweight) should be viewed as cholesterol-dependent atherosclerotic risk factors and not in themselves as atherogenic. There is no doubt that atherosclerotic events are more common in people with these risk factors, but only in those populations with an average serum total cholesterol level above 3.9 mmol/l. The only absolute, unequivocal, independent atherosclerotic risk factor is an elevated serum total or, more specifically, low density lipoprotein (LDL)-cholesterol level. Whether a low level of high-density lipoprotein cholesterol is an independent risk factor is not clear, but it should probably be regarded as an additive risk when the serum LDL-cholesterol is elevated.
_
The luminal narrowing due to plaque formation or precipitating thrombus in atherosclerosis results in adverse cardiac events (myocardial infarction, angina), brain injury (ischemic stroke) and peripheral vascular disease. Coronary artery disease (CAD) is the most common of all these, resulting in myocardial infarction and angina pectoris. An increased serum level of low-density lipoprotein (LDL) is sufficient to induce the atherosclerotic changes. The facilitating factors such as smoking, hypertension, diabetes mellitus, male sex, and family history or genetic susceptibility further add nuances to the disease presentation. Physical inactivity increases Oxidative Stress, Endothelial Dysfunction, platelets activation and atherosclerosis. Depending on the clinical symptom, the atherosclerotic plaques may be asymptomatic (subclinical disease), obstructive (stable angina, transient ischemic attack, amaurosis fugax), and symptomatic (acute thrombosis leading to acute coronary syndrome, stroke).
______
Pathophysiology of Atherosclerosis:
The endothelial cell layer maintaining vascular homeostasis. Endothelial cells provide a functional link between blood in the lumen and the vessel wall. They transduce many physiological stimuli, that have effects on blood cells, endothelium itself and on neighbouring smooth muscle cells. Nitric oxide (NO) is one of the most important signaling molecules produced by the endothelium. NO was originally identified as ‘endothelium-derived relaxing factor’. In normal blood vessels, NO is generated by the enzyme endothelial nitric oxide synthase (NOS), which is present in endothelial cells.
Endothelial dysfunction in Atherosclerosis:
The overall effects of NO on the vessel wall are to inhibit the processes that contribute to early atherosclerosis. İn diseased artery, early and characteristic feature of is abnormal function of the endothelium (endothelial dysfunction); characterized by loss of endothelial NO bioactivity and associated activation of the endothelium; resulting in predisposition to thrombosis and inflammatory cell adhesion and recruitment. İn the following conditions, endothelial dysfunction can be detected before macroscopic atherosclerosis is visible:
- Hypercholesterolemia
- Smoking
- Hypertension
- Heart failure.
İn all these patients with atherosclerosis, arteries are structurally normal. Clinically, endothelial dysfunction manifests as abnormalities in NO-mediated vascular responses: (for example: lack of dilatation, or paradoxical constriction of coronary artery segments in response to intracoronary infusion of acetylcholine).
_
As a result of endothelial dysfunction, lipid and cholesterol continuously accumulate within the intima from a young age and recruit inflammatory cells into the lesion. Atherosclerosis gradually progresses from intimal thickening into fatty streak and foam cell formation and eventually into a cholesterol rich fibrous plaque which can narrow or block the arterial lumen, hampering the blood flow. The formation of an unstable plaque with characteristic fibrous cap thinning, a necrotic core and the excess infiltration of inflammatory cells, increases the risk of plaque rupture. The formation of new blood vessels in atherosclerosis has been of growing interest over the last 20 years since there is a great deal of uncertainty into whether it plays a beneficial or detrimental role in this disease. Angiogenesis is thought to contribute to atherosclerosis and plaque instability.
_
Epidemiological studies have shown that atherosclerosis can be initiated by a number of different conditions such as hypertension, diabetes, and lifestyle factors such as lack of exercise, heavy alcohol consumption, smoking and being overweight. The most common risk factor for atherosclerosis is hypercholesterolemia. Dietary lipids are transported in the blood as chylomicrons whereas endogenous lipids are transported as Very Low Density Lipoproteins (VLDL), Low Density Lipoproteins (LDL) and High Density Lipoproteins (HDL). High levels of blood LDLs and VLDLs are associated with the increased risk of atherosclerosis as they carry cholesterol to peripheral tissues. HDLs are responsible for removal of cholesterol from peripheral tissues and therefore low levels are observed in atherosclerosis. These risk factors can be controlled, however, there are also factors which increase susceptibility to atherosclerosis such as gender, genetic susceptibility and age which cannot be controlled. Although, the presence of atherosclerotic lesions deep within the intima can occur from a young age, atherosclerosis is associated with aging as symptoms of the disease do not usually manifest until later in life.
_____
Low-density lipoprotein (LDL) is conventionally quantified in terms of the mass of cholesterol carried by these particles. LDL cholesterol (LDL-C) has been the standard measure of LDL and LDL-attributable cardiovascular disease (CVD) risk for so long that “LDL” and “LDL-C” tend to be used interchangeably. However, the two terms are not synonymous because the cholesterol content of LDL particles varies more than 2-fold among individuals. One person may have large, more cholesterol-rich LDL while a second may have smaller cholesterol-poor LDL particles. At the same LDL-C concentration, the second person will have higher numbers of LDL particles. A priori, it is not clear whether the cholesterol in LDL (LDL-C) or the number of LDL particles would be the more informative marker of LDL-attributable atherosclerotic risk. On the one hand, a more cholesterol-rich LDL particle deposits more cholesterol in the artery wall and from this perspective may be considered more atherogenic than a cholesterol-poor particle. On the other hand, the probability that a particle’s cholesterol will be delivered to an atheroma depends largely on particle number: how many LDL particles enter the artery wall, become oxidized, and are finally taken up by macrophage foam cells. Lipid profile test done in laboratory typically shows LDL cholesterol.
_____
Cholesterol crystal induced arterial inflammation and destabilization of atherosclerotic plaque, a 2015 review:
Accumulation of low-density lipoprotein in the sub-intima provides esterified cholesterol (ESC) to macrophages and smooth muscle cells that convert it into free cholesterol (FRC) by cholesteryl ester hydrolases (CEHs). Membrane-bound cholesterol carriers transport FRC to high-density lipoprotein (HDL). Impaired HDL transport function and altered composition can lead to extracellular accumulation of FRC, whereas impaired membrane carrier activity can lead to intracellular FRC accumulation. Saturation of FRC can result in cholesterol crystallization with cell death and intimal injury. Disequilibrium between ESC and FRC can impact foam cell and cholesterol crystal (CC) formation. Cholesterol crystals initiate inflammation via NLRP3 inflammasome leading to interleukin-1β (IL-1β) production inducing C-reactive protein. Eventually, crystals growing from within the plaque and associated inflammation destabilize the plaque. Thus, inhibition of inflammation by antagonists to IL-1β or agents that dissolve or prevent CC formation may stabilize vulnerable plaques.
_
Figure below shows cholesterol transport within cells and extracellular space:
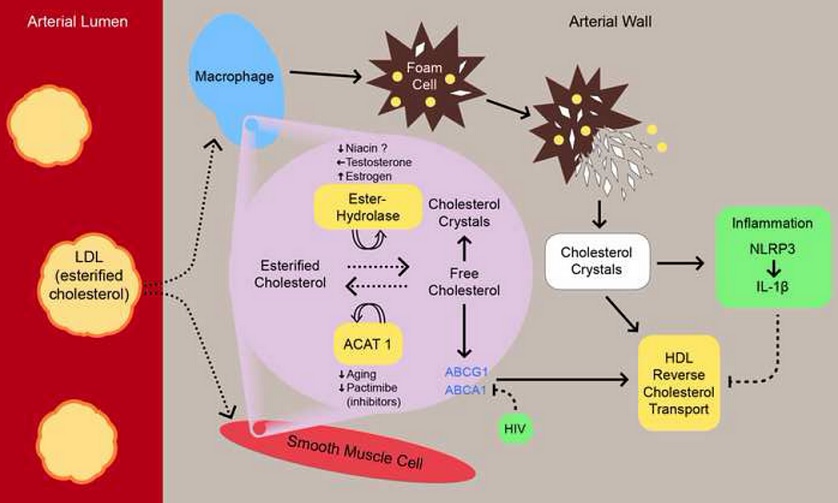
Equilibrium between esterified and free cholesterol is noted with membrane transporters driving free cholesterol into the extracellular space where it is taken up by high-density lipoprotein. Dying foam cells overloaded with esterified cholesterol and crystals release their content into the extracellular space. Free cholesterol build-up in the extracellular space leads to crystallization. ABCA1, ABCG1, ATP-binding cassette A-1, G-1; ACAT 1, acyl-coenzyme A cholesterol acyltransferase 1; HDL, high-density lipoprotein; HIV, human immunodeficiency virus; IL-1β, interleukin-1β; LDL, low-density lipoprotein; NLRP3, NLRP3 inflammasome.
_
High-density lipoprotein functionality and free cholesterol accumulation:
High-density lipoprotein reverse transport of cholesterol out of arterial wall cells requires the conversion of ESC to FRC so that it can be mobilized via membrane-bound transporters, ATP-binding cassette A-1 and G1 (ABCA-1, ABCG-1). A failure of this pathway will lead to both ESC and FRC build-up. Moreover, HDL has been shown to dissolve CC directly. Thus, with dysfunctional HDL, FRC will accumulate in both the cell membrane and the extracellular space promoting the formation of CC. Of note, one of the conditions leading to HDL dysfunction is inflammation. As atherosclerosis builds in the arterial wall, there is a rise in both local and systemic inflammation that can induce HDL dysfunction leading to a vicious positive feedback cycle that favours atherosclerosis build-up with further worsening of HDL function.
_
Frequency of Cholesterol Crystals in Culprit Coronary Artery Aspirate During Acute Myocardial Infarction and Their Relation to Inflammation and Myocardial Injury, a 2017 study:
Cholesterol crystals (CCs) have been associated with plaque rupture through mechanical injury and inflammation. This study evaluated the presence of CCs during acute myocardial infarction (AMI) and associated myocardial injury, inflammation, and arterial blood flow before and after percutaneous coronary intervention. Patients presenting with AMI (n = 286) had aspiration of culprit coronary artery obstruction. Aspirates were evaluated for crystal content, size, composition, and morphology by scanning electron microscopy, crystallography, and infrared spectroscopy. These were correlated with inflammatory biomarkers, cardiac enzymes, % coronary stenosis, and Thrombolysis in Myocardial Infarction (TIMI) blush and flow grades. Crystals were detected in 254 patients (89%) and confirmed to be cholesterol by spectroscopy. Of 286 patients 240 (84%) had CCs compacted into clusters that were large enough to be measured and analyzed. Moderate to extensive CC content was present in 172 cases (60%). Totally occluded arteries had significantly larger CC clusters than partially occluded arteries (p <0.05). Patients with CC cluster area >12,000 µm2 had significantly elevated interleukin-1 beta (IL-1β) levels (p <0.01), were less likely to have TIMI blush grade of 3 (p <0.01), and more likely to have TIMI flow grade of 1 (p <0.01). Patients with recurrent AMI had smaller CC cluster area (p <0.04), lower troponin (p <0.02), and IL-1β levels (p <0.04). Women had smaller CC clusters (p <0.04). Macrophages in the aspirates were found to be attached to CCs. Coronary artery aspirates had extensive deposits of CCs during AMI. In conclusion, presence of large CC clusters was associated with increased inflammation (IL-1β), increased arterial narrowing, and diminished reflow following percutaneous coronary intervention.
_
Incidence, factors, and clinical significance of cholesterol crystals in coronary plaque: An optical coherence tomography study, 2019:
Investigators analyzed 340 individuals to explore the incidence, factors, and clinical consequence of cholesterol crystal (CC) in coronary plaque. They observed no significant variation in baseline clinical characteristics between individuals with and without CC except for eicosapentaenoic acid/arachidonic acid ratio and hemoglobin A1c levels. Among individuals with versus without CC, the incidence of major adverse cardiovascular events (MACE) and non-target vessel revascularization was greater at 1-year, and the presence of CC was significantly correlated with a greater rate of 1-year MACE.
_______
Foam cell and fatty streak formation:
As a result of endothelial dysfunction, lipids accumulate deep within the intima of the artery wall in the first decade of atherosclerosis. This results in foam cell and fatty streak formation. LDLs enter the intima through the endothelium and bind to the extracellular matrix. Due to the decrease in nitric oxide production or activity, LDLs are the oxidized by enzymes such as nitric oxide synthase (NOS), 15-lipoxygenase (15-LO) or by reactive oxygen species (ROS). As a result of enzyme oxidation, ‘minimal’ modification (mm) LDLs and oxidized LDLs are formed. MmLDLs induce endothelial cells to express adhesion molecules such as P-selectin, E-selectin, intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1) which are essential for leukocyte transmigration, interactions with inflammatory cytokines as well as the formation of neovessels. Monocytes from the blood attach to the endothelium via adhesion molecules and transmigrate to enter the sub-endothelial space. Monocytes differentiate into macrophages after migration and express scavenger receptors enabling the uptake of oxidized LDLs. The accumulation oxidized LDLs in macrophages leads to the formation of foam cells which contribute to the formation of fatty streaks. Foam cell cholesterol is esterified and stored as lipid droplets or extruded out of the cell through cholesterol transporters such as ABC-A1 and accepted by HDLs. Fatty streaks have a yellow discoloration due to the accumulation of foam cells and represent the bulk of the atherosclerotic lesion, occupying up to 6 layers of the intima of the artery wall. In early atherosclerotic lesions, the distribution of fatty streaks in the vasculature is predominantly in areas where there are changes in blood flow such as bifurcations and back currents causing endothelial injury.
_____
Intermediate lesion and atheroma:
The third decade of atherosclerosis involves the formation of an intermediate lesion and atheroma. Type 2 T helper lymphocytes stimulate foam cells to secrete cytokines which leads to smooth muscle cells migration from the medial part of the vascular wall into the intima. Migrated muscle cells may take up lipid droplets, proliferate and secrete fibrin, collagen and proteoglycans which make up a poorly developed extracellular matrix. The combination of these extracellular proteins and fibrinogen from the blood leads to the formation of a fibrous plaque. Type 1 T helper lymphocytes release CD40 and interferon-γ which are both involved in inflammation within the atherosclerotic lesion.
_
Briefly, oxidized LDL enhances the oxidative stress in the intima which leads to activation of inflammatory cascade involving inflammatory receptors [receptor for advanced glycosylation end products (RAGEs), toll-like receptors (TRLs), and triggering receptor expressed on myeloid cells (TREMs)], downstream signaling kinases [protein kinase C (PKCs), c-Jun NH2-terminal kinase (JNK), ERK, mitogen-activated protein kinase (MAPK) etc.] and pro-inflammatory cytokines [interleukin (IL)-6, tumor necrosis factor (TNF)-α]. This leads to increased monocytic infiltration, M1 macrophage predominance and foam cell formation, which further enhances the inflammation and vascular smooth muscle cells (VSMCs) proliferation resulting in atherosclerosis. Further, research studies have also demonstrated the involvement of inflammatory surface markers (TREMs and TLRs) and pro-inflammatory cytokines (IL-6 and TNF-α) in plaque vulnerability.
_
Fibrous plaque formation:
The fibrous plaque consists of an accumulation of smooth muscle cells suspended in a matrix of collagen and some elastic fibers, and surrounded by a dense fibrous cap. The smooth muscle cells adopt a lacunar-shape in dense layers of collagen in the basement membrane. Plaque contains cholesterol & other fatty materials, calcium, and blood components that stick to the artery wall lining.
During the progression of atherosclerosis, endothelial cells, macrophages, and smooth muscle cells die from apoptosis or necrosis. Disintegration of foam cells, loss of smooth muscle cells, and production of matrix metalloproteinases by activated leukocytes have detrimental consequences—leading to the formation of destabilizing lipid-rich cores and fragile and rupture-prone fibrous caps.
_______
Figure below shows evolution of human atherosclerosis.
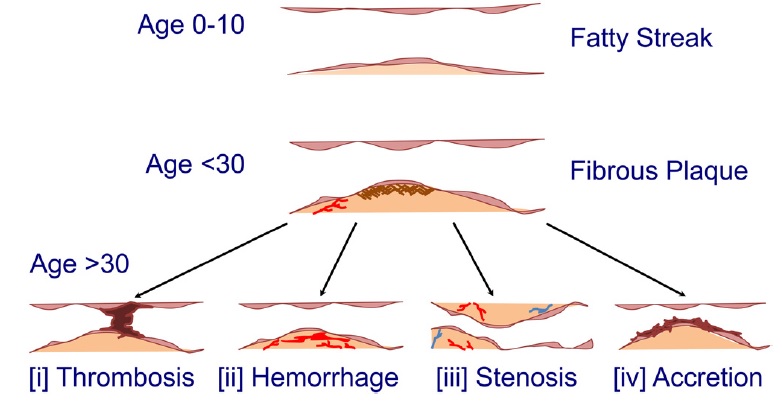
Fatty streaks represent one of the earliest visible lesions in atherogenesis, and have been observed early in life. These fatty streaks often evolve into fibrous plaques, coupled with intimal hyperplasia and accompanying changes to the extracellular matrix. As these fibrous plaques further grow, they may progress toward several outcomes depending on microenvironmental and environmental factors:
- occlusive thrombosis, as a result of fibrous cap rupture because of the combination of endothelial cell loss revealing the prothrombotic basal lamina, degradation of the fibrous cap as a result of proteases or proteinases or possibly through volume expansion and puncture as a result of cholesterol crystals;
- intraplaque hemorrhage, as a result of abnormal neovascularization, vascular permeability, or increasing plaque volume;
- stenosis, as a result of progressive luminal reduction by plaques leading to further lipid-richness, fibrosis, and “hardening” of the arteries; or
- mural thrombotic accretion, as a result of excess subendothelial accumulation of fibrin and platelets.
______
The American Heart Association categorized atherosclerotic plaques into 6 different types (table below) of lesions based on the characteristic components and pathogenic mechanisms of the various advanced atherosclerotic lesions.
| Stage | Features |
| I | Adhesion of monocytes to the intact endothelial surface and their migration into the intima |
| II | Fatty streak. A focal accumulation of lipid-filled macrophages (foam cells) in the intima. The overlying endothelium is intact.
Some T-lymphocytes are also present |
| III | Extracellular lipid in addition to that contained within foam cells, and smooth muscle cell numbers have increased |
| IV | Extracellular lipid coalescing into the centre of the plaque and a layer of smooth cells |
| Va | Fully formed lipid core with an acellular mass of cholesterol, some of which is in the crystalline form, and well-developed cap of fibrous tissue separating the core from the lumen |
| Vb | Same features of Va but also calcification |
| Vc | Solid and fibrous plaques without a lipid core and a very minor macrophage content |
| VI | Plaques complicated by thrombosis |
_
Modified American Heart Association classification based on morphological description:
| Description | Thrombosis | |
| Non-atherosclerotic intimal lesions | ||
| Intimal thickening | The normal accumulation of smooth muscle cells (SMCs) in the intima in the absence of lipid or macrophage foam cells | Absent |
| Intimal xanthoma, or “fatty streak” | Luminal accumulation of foam cells without a necrotic core or fibrous cap. Based on animal and human data, such lesions usually regress. | Absent |
| Progressive atherosclerotic lesions | ||
| Pathological intimal thickening | SMCs in a proteoglycan-rich matrix with areas of extracellular lipid accumulation without necrosis | Absent |
| Erosion | Luminal thrombosis; plaque same as above | Thrombus mostly mural and infrequently occlusive |
| Fibrous cap atheroma | Well formed necrotic core with an overlying fibrous cap | Absent |
| Erosion | Luminal thrombosis; plaque same as above; no communication of thrombus with necrotic core | Thrombus mostly mural and infrequently occlusive |
| Thin fibrous cap atheroma | A thin fibrous cap infiltrated by macrophages and lymphocytes with rare SMCs and an underlying necrotic core | Absent; may contain intraplaque haemorrhage/fibrin |
| Plaque rupture | Fibroatheroma with cap disruption; luminal thrombus communicates with the underlying necrotic core | Thrombus usually occlusive |
| Calcified nodule | Eruptive nodular calcification with underlying fibrocalcific plaque | Thrombus usually non-occlusive |
| Fibrocalcific plaque | Collagen-rich plaque with significant stenosis usually contains large areas of calcification with few inflammatory cells; a necrotic core may be present | Absent |
________
The atherosclerotic plaque development in a nutshell:
-Endothelial cell dysfunction is the earliest detectable physiologic manifestation of atherosclerosis. The major atherogenic risk factors, such as smoking, high low-density lipoprotein (LDL) levels, hypertension, and diabetes, have all been shown to impair endothelial function.
-Normal endothelium has antithrombotic, antiinflammatory, and vasomodulatory functions through secretion of substances, such as prostacyclin and nitric oxide (NO), which inhibit platelet activation and promote vasodilatation. NO inhibits expression of the adhesion molecules responsible for inflammatory cell recruitment. -Both the barrier function and secretory capacity of the endothelium are disrupted in atherosclerosis.
-This manifests as an increase in permeability to lipids and inflammatory cells (mainly monocytes and T lymphocytes) derived from the blood.
-The combination of endothelial dysfunction and high circulating levels of atherogenic lipoproteins leads to the accumulation of lipid-laden, monocyte- derived foam cells in the subendothelial layer, forming the early atherosclerotic lesion.
– Accumulation of foam cells and their subsequent death produces an acellular core of cholesterol esters and cell debris.
-Vascular smooth muscle cells (VSMCs) migrate from the medial layer of the vessel and synthesize extracellular matrix components, such as elastin and collagen, to form the fibrous cap.
-The fibrous cap contains inflammatory cells, predominantly macrophages, but also some T lymphocytes and mast cells.
– Intimal thickening is the first and most common clinically detectable manifestation of atherosclerosis in humans.
______
Atherosclerotic plaques are asymmetric focal thickenings of the intima due to accumulation of varying quantities of foamy macrophages, blood products, smooth muscle cells, lipids, collagen, necrotic debris, and calcium. Hence, imaging tools used for the investigation of atherosclerotic plaque would optimally not only outline the boundaries of atherosclerotic lesions but also differentiate various components within the plaque to fully characterize the lesion. In coronary imaging, the ultimate goal is to identify individuals at increased risk for acute coronary syndromes (ACSs), a frequent reason for hospitalization, sudden death, and chronic disease. Because ACSs are often the first manifestation of disease in previously asymptomatic individuals, early identification of patients at risk for plaque rupture has become an important goal in primary prevention.
______
Stable Plaques:
Although stability of a plaque is not predictable, calcified and fibrotic plaques (fibrocalcific) are associated with greater clinical stability. Silent plaque rupture is a common process and, similar to wound healing, results in contraction, loss of adaptive remodeling, and progressive luminal stenosis. In patients with stable angina, including those with history of a coronary artery bypass graft, there is a substantial variability in plaque morphology. This group is probably the most commonly referred for imaging, including coronary CT angiography (CTA). In this group, fibrous and calcified plaques are common, and it is not unusual to find lesions with greater than 75% luminal stenosis. On the other hand, lipid-rich lesions and luminal thrombosis are unusual.
The amount of coronary calcification is proportional to plaque burden and stenosis severity and increases with aging, especially in men. Microcalcifications (speckled) are common in ruptured, erosive, and TCFA but are not detectable in routine imaging studies. However, it is not rare to see larger fragmented or diffuse calcifications in ruptured plaques, TCFA, and healed plaques, but it is an unusual finding in erosive plaques. Therefore, absence of calcification in a lesion with large necrotic core may be more suggestive of plaques with superficial erosion.
Calcified nodules are lesions with the greatest amount of calcification relative to plaque area and only rarely trigger thrombosis. Calcified nodules are generally located in the proximal end of the left anterior descending (LAD) and left circumflex arteries, whereas calcified nodules within the right coronary arteries are evenly and more distally distributed. It has been shown on CT that shelllike and diffuse calcifications are more frequently associated with 50% or greater stenosis than those with punctate calcified plaques.
_____
From stable atherosclerotic plaque to Vulnerable plaque:
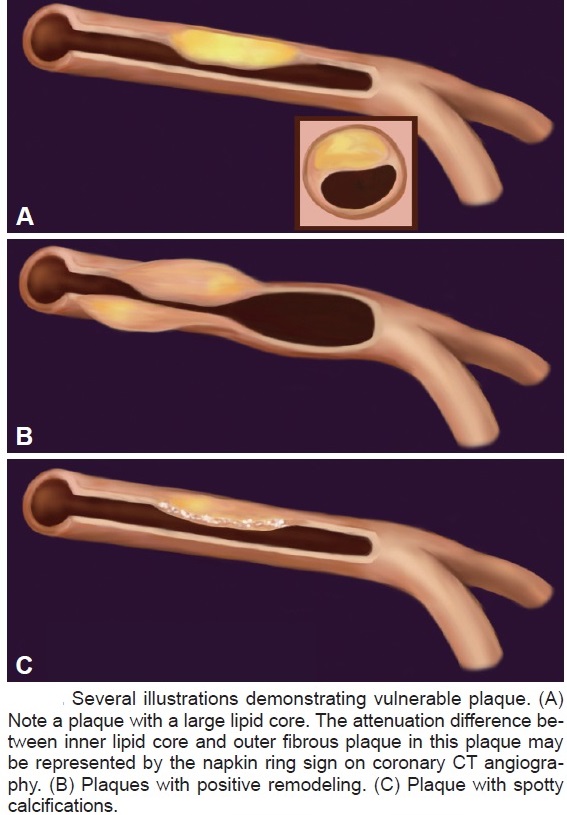
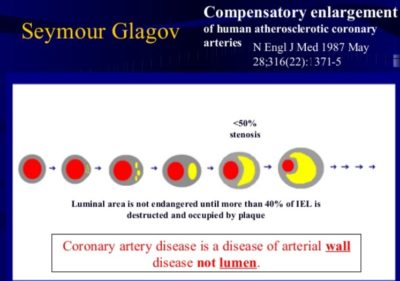
The “vulnerable plaque” was firstly defined as a non-obstructive, silent coronary lesion that suddenly becomes obstructive and symptomatic. It is responsible for the majority of acute coronary events. The most complete definition of this phenomenon was proposed in 2003 by Naghavi et al., reporting that vulnerable plaques are not only susceptible to rupture, but they are more broadly susceptible to thrombosis or may rapidly progress to a culprit lesion. On the basis of large retrospective autopsy studies, major and minor functional criteria were defined to potentially assess the degree of vulnerability of a plaque. Some atherosclerotic plaque features are particularly associated to vulnerability: structural characteristics (increased lipid content, between 10 and 25%, the presence of a thin cap [<65 μm]), inflammatory cellular accumulation (activated macrophages), presence of positive remodelling of the coronary vessel and neovascularisation. The size of the necrotic core and the thickness of the fibrous have been indicated as major structural determinants of vulnerability. These aspects make the cap of a plaque less resistant to the circumferential wall stress. The composition of the lipid necrotic core of a vulnerable plaque is characterised by an increased free/esterified cholesterol ratio, resulting in a higher risk of rupture and thrombosis. The high lipid content could be the reason for the yellow color of this type of plaque. Inflammatory cell infiltration in a vulnerable plaque is particularly capable of degrading the extracellular matrix by secretion of proteolytic enzymes. Their activation is also related to a higher risk of intraplaque hemorrhage (IPH) that is one of pivotal event in plaque disruption. As shown in a rabbit model, the accumulation of erythrocyte membranes within the lesion may increase the risk of plaque destabilisation by contributing to the deposition of free cholesterol, macrophage infiltration, and enlargement of the necrotic core. Another possible mechanism is that when a red blood cell (RBC) is damaged it releases its iron content which can activate the reactive oxygen species (ROS) thereby increasing the plaque vulnerability for rupture. A vulnerable plaque has been shown to stimulate a vessel’s compensatory remodelling due to the digestion of the internal elastic lamina, which results in an eccentric growth of the atheroma and an outward enlargement of the vessel wall without luminal compromise. Historically, the most important diagnostic technique for studying atherosclerotic disease was to determine the residual luminal diameter by angiographic measurement of the stenosis. However, it has become clear that vulnerable plaque rupture as well as thrombosis, rather than stenosis, triggers most acute ischaemic events and that the quantification of risk based merely on severity of the arterial stenosis is not sufficient. Therefore, in the last few decades a great deal of progress has been made in the field of techniques that allow us to study arterial wall morphology, plaque composition and its inflammation, and metabolic processes. Despite some limitations due to the fact that a “histologically” defined vulnerable plaque might not clinically rupture, a gold-standard imaging technique should check each one of these structural characteristics to identify a vulnerable plaque. This technology should have high resolution in order to measure the cap’s thickness. It should also allow identifying and quantifying the cap’s cholesterol and the composition of the inflammatory cells, the presence of IPH, the iron content and the positive vessel remodelling. Since the composition of the high-risk plaques varies, depending on their anatomical size, with striking heterogeneity even within the same individual, non-invasive imaging modalities that are able to detect and characterise atherothrombotic lesions in their various stages and their different anatomical regions are clinically needed. Non-invasive techniques might allow us to rapidly screen low-moderate risk patients and therefore to prevent acute coronary syndrome (ACS). On the other hand, invasive techniques that have high microscopic resolution are needed to study the plaque composition during angiography in patients with known atherosclerotic disease.
Although a large lipid core is associated with plaque vulnerability and myocardial infarction, there is no area threshold above which a plaque becomes unstable. Using simulations, it has been shown that plaques are more prone to rupture in the early stages of positive remodeling and that necrotic core thickness rather than area determines plaque stability. Positive remodeling (outward expansion) is defined as 5% increase in the luminal cross-section at the site of plaque compared with the normal proximal segment of the vessel (remodeling index > 1.05). Positive remodeling indicates plaque instability and is common in plaques with large necrotic cores, TCFA, and hemorrhage. It is not seen in chronic stable fibrotic plaques. In fact, stable lesions may reveal negative remodeling (< 0.95). Unfortunately, not all vulnerable plaques display positive remodeling—especially erosive plaques usually show no or little positive remodeling.
_______
Figure below shows pathogenesis of plaque formation and plaque rupture.
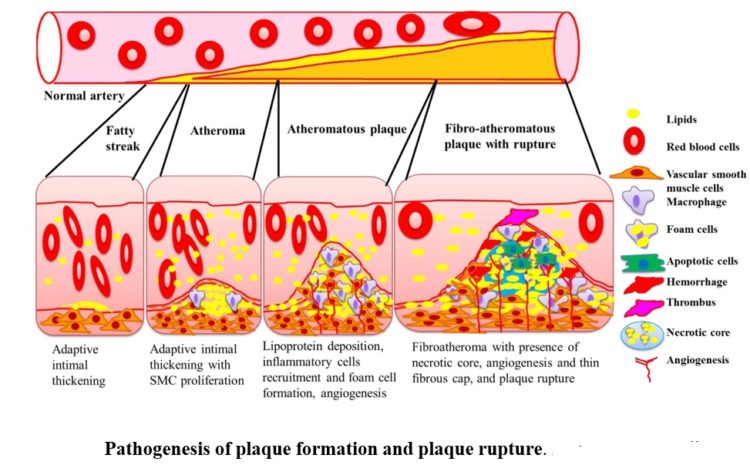
Deposition of the lipids in the intima leads to fatty streak formation and adaptive intimal thickening. Infiltration of inflammatory cells, lipid deposition, and vascular smooth muscle cells (SMCs) proliferation results in progression of the fatty streak to atheroma and atheromatous plaque. Formation of the necrotic core due to increased apoptosis and necrosis in plaque; increased lipid deposition and angiogenesis and thinning of the fibrous cap results in the development of vulnerable fibroatheromatous plaque. The precipitating factor for acute coronary syndrome (ACS) is luminal thrombus or a sudden plaque hemorrhage within the atherosclerotic plaque. The ACS is not necessarily accompanied with concomitant vasospasm. Plaque rupture is the most frequent cause of thrombosis. Plaque rupture results in the exposure of highly thrombogenic, red cell–rich necrotic core material to the blood.
_
Average composition of advanced coronary plaque:
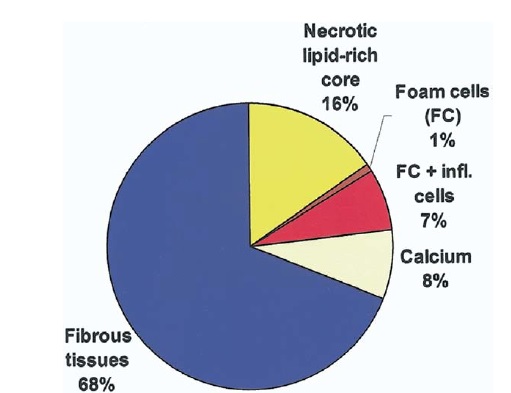
Figure above is pie diagram illustrating the average composition of advanced atherosclerotic plaques (75% stenosis by histology) in the coronary artery tree in fatal myocardial infarction. Hypocellular tissues (fibrosis, calcium, and necrosis) constitute by far the most voluminous plaque components.
______
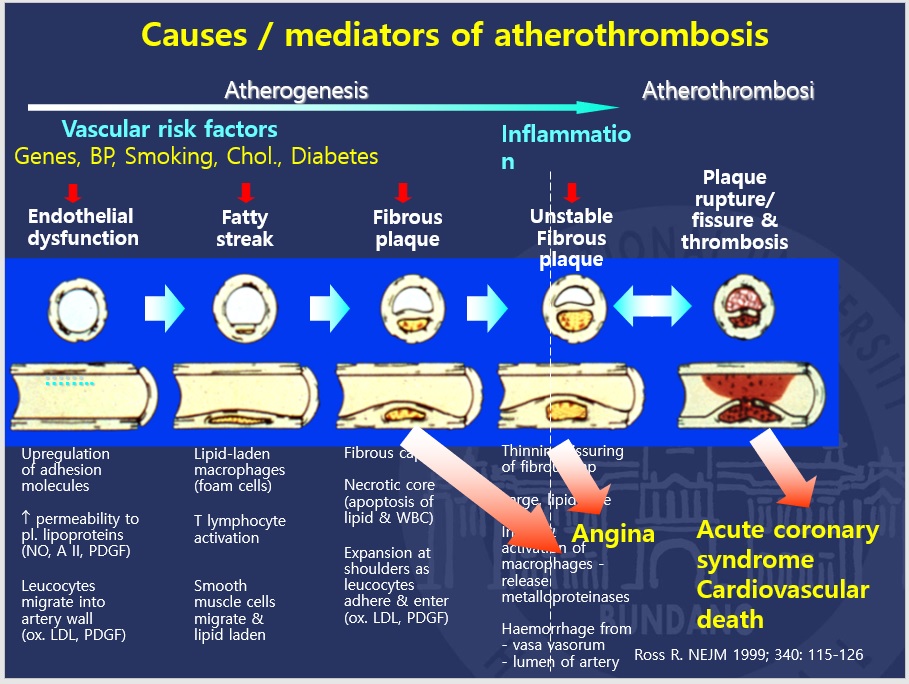
Evolution of atherosclerosis to thrombosis i.e. atherothrombosis:
_
_
Figure below shows development of atherosclerosis and progression to thrombosis and clinical events.
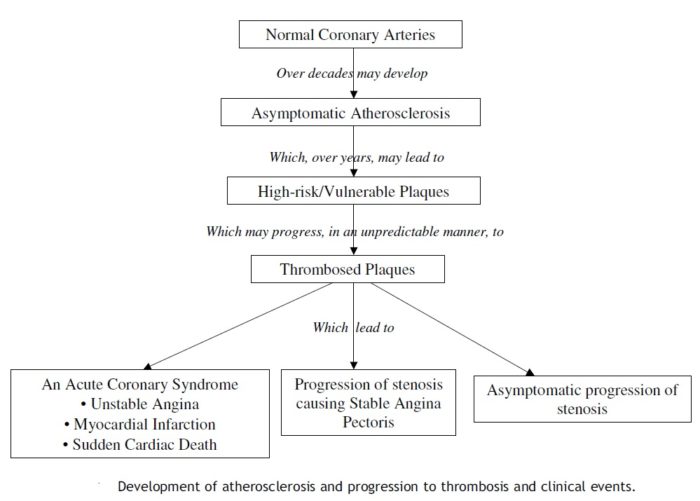
The progression from asymptomatic atherosclerosis, to a high-risk/vulnerable plaque, to a thrombosed plaque, and to clinical events is presented in the figure above. It is of note that the later stages of the progression may be repeated in a relatively short time interval as documented by the high short-term risk of a recurrent event in patients with acute coronary syndromes. This may be caused by rethrombosis of the lesion causing the index event, and/or the simultaneous occurrence of multiple high-risk/vulnerable plaques and/or thrombosed plaques, that have not previously caused symptoms. An acute coronary syndrome may be a clinical marker of widespread (multifocal) disease activity in the coronary arteries, possibly related to inflammation.
_______
Atherosclerosis is a chronic degenerative dynamic disease of the arterial wall, which mainly involves large and medium-sized systemic arteries. Clinical ischaemic symptoms, such as angina pectoris, usually appear when an increase in myocardial oxygen requirements leads to a transitory imbalance between supply and demand (reduced supply and increased demand). This pathophysiological event is often caused by a coronary plaque that realizes a flow-limiting stenosis reducing tissue perfusion. If this clinical situation is constant, coronary atherosclerotic plaques have been described as stable lesions of fibrotic morphology. Atherosclerosis may, however, take on other clinical forms due to a sudden transition in clinical instability, causing unheralded events, such as myocardial infarction and stroke. The transition from asymptomatic (non-obstructive) to symptomatic disease is mainly related to a sudden atherosclerotic plaque rupture and subsequent thrombosis. These “vulnerable” lesions might be also non-obstructive plaques. Atherosclerotic plaque rupture occurs as a result of the interactions between external mechanical triggers and vulnerable regions of the plaque when forces acting on the plaque exceed its tensile strength. Even if part of the factors leading to stress on the plaque cap have been identified (i.e. blood pressure, excursion between systolic and diastolic pressure and arterial compliance) and specific intervention have been designed, many of these external forces is still unknown and therefore, it is presently difficult to design effective treatments to prevent plaque rupture. However, plaque tissue properties undoubtedly determine the mechanical strength of plaques and may be a realistic target for therapeutic intervention, thereby decreasing the incidence of heart attack and stroke. Because of the central role of inflammation and hypercoagulability in the genesis and progression of a vulnerable plaque, a variety of circulating biomarkers has been identified. Although their clinical usefulness in the early diagnosis of acute myocardial infarction (AMI) has not been demonstrated yet, the measurement of inflammatory biomarkers of plaque vulnerability (such as myeloperoxidase [MPO], C-reactive protein [CRP], interleukin [IL]-6), or CX3CL1/fractalkine) and of blood markers reflecting thrombogenic activity (fibrinogen, D-dimer, soluble (s)CD40L, sP-selectin and activated platelets) could provide some incremental information regarding the risk stratification of patients.
______
Changes observed routinely in the coronary arteries in fatal ischemic heart disease (IHD) are reviewed:
- The coronary arteries are diffusely involved by atherosclerotic plaques. Although the lumens of some segments are more severely narrowed than others, all portions of the extramural coronary tree are involved by the atherosclerotic process.
- In fatal IHD, with rare exception, the lumens of at least two of the three major coronary arteries are >75% narrowed by old atherosclerotic plaques. The most severe narrowing tends to be in the more proximal portions of the left anterior descending and left circumflex branches; the distal half of the right coronary artery is prone to narrowing that is as severe as that in its proximal portion.
- The atherosclerotic process is limited to the epicardial coronary arteries, i.e., the major trunks and their near right-angle branches. The intramural (intramyocardial) coronary arteries are spared by the atherosclerotic process.
- The coronary artery responsible for perfusing with oxygen the area of myocardial ischemia is not necessarily the most severely narrowed of the 3 major coronary arteries but its lumen is virtually always >75% narrowed at some point by atherosclerotic plaques.
_____
Is coronary artery more prone to atherosclerosis than other arteries?
The coronary arteries are, in fact, more prone to blockages than many other arteries in the human body. Coronary arteries are prone to blockages because they’re smaller vessels, delicate vessels and fewer in number supplying vital organ. Blood flow to heart through coronaries is 215 ml/minute at rest and rises to 750 ml/minute during exercise. For this amount of blood flow, the size of coronary arteries is small. Blood flow to brain is 650ml/minute at rest but size of carotid and vertebral arteries are comparatively far larger. Another reason is that there is to-and-fro blood flow in the coronary arteries, as well as in the legs and the carotid arteries, two other regions prone to blockages. This turbulent blood flow hurts the lining of arteries, much like heavy winds blowing back and forth over time can take a toll on the trees in a forest. Where blood flow resembles a gentle breeze in one direction, as in your arms, blockages are less likely to occur.
The story is probably even more complicated than disturbed blood flow, because some blood vessels appear to be more resistant to disease regardless of the flow pattern. This is particularly true for the left internal mammary artery, an artery that runs down the inside of the chest wall. This artery is unusually resistant to blockages, one of the reasons it is a standard artery used in coronary bypass surgery. Human atherosclerotic lesions in saphenous vein bypass grafts are vulnerable and have a higher risk to disrupt than native atherosclerotic lesions (de Vries et al., 2016, Yahagi et al., 2016).
Is left coronary system more susceptible to atherosclerosis than right?
On the basis of pathological, angiographical, intravascular ultrasound and computed tomography data coronary atherosclerosis appears to be more prevalent in the left coronary arterial system compared to the right. However, the pathophysiological mechanisms implicated in this discrepancy largely remain uncertain. The hemodynamic or anatomical differences between the right and left coronary artery might play a key role. Physiologically, the right coronary flow is more uniform during the cardiac cycle compared to the left, which experiences a remarkable systolic decline accompanied by a significant diastolic increment. Thus, the oscillatory shear stress, that constitutes a proved atherogenic factor, is more intense in regions with disturbed flow in the left coronary system. Likewise, the wall stress is more oscillatory during the cardiac cycle in the left coronary artery. On top of that, several differences regarding the anatomical configuration (3D geometry, branching) and the phasic motion between the right and the left arterial system appear to play a critical role in the modulation of the local atherogenic environment.
_______
Underlying causes of Sudden Fatal and Nonfatal Cardiac Events:
Figure below delineates the underlying causes of acute cardiac events. The first branch point of the tree indicates patients who lack significant atherosclerosis or related myocardial damage, that is, those who have no ischemic heart disease. This leaves the patients with atherosclerosis, some of whom also have a hypercoagulable state. The next branch point involves the presence or absence of an occlusive or subocclusive thrombus. A thrombus identifies a culprit plaque that may be ruptured or nonruptured.
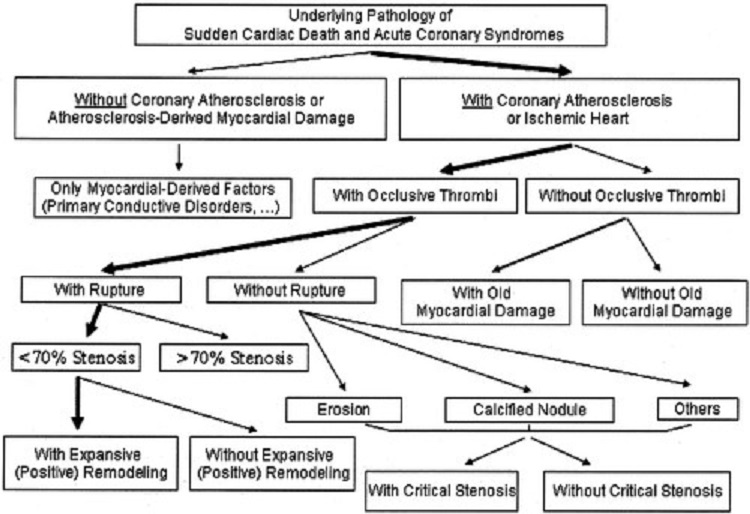
Figure above delineates underlying pathology of acute coronary syndrome, (i.e., unstable angina, acute myocardial infarction and sudden cardiac death).
_____
Clinical Presentation vis-à-vis plaques:
Myocardial ischaemia result due to acute or persistently (chronic) decrease myocardial blood flow due to narrowed coronary lumen diameter, mainly caused by coronary Atherosclerosis.
Mechanisms:
- When patient has stable coronary obstruction due to significant plaque stenosis ( ≥ %50 lumen diameter narrowing) they mostly remain asymptomatic in the resting conditions. At this situation coronary flow never respond (augmented) to increased myocardial oxygen demand (strenuous exertion, emotional stress); so myocardial ischaemia occurs as a result of demand supply mismatch; clinically termed Chronic Coronary artery disease (CAD) or stable angina pectoris.
- On the other hand, patient who has previously near normal coronary flow with insignificant obstructive plaque (<%50 luminal narrowing); when coronary flow suddenly decrease below the critical level (supply not matching resting demand) from result of abrupt thrombosis superimposed over plaque disruption, acute myocardial ischeamia develops resulting in Acute coronary syndromes (ACS). Acute coronary syndrome (ACS) commonly results from rupture of thin-cap fibroatheroma (TCFA), and occasionally results from erosion or calcified nodules. Main clinical presentations of ACS are sudden cardiac death, symptomatic or asymptomatic acute myocardial ischaemia/infarction. ACS can also result from sudden thrombotic occlusion of significant plaque stenosis ( ≥ %50 lumen diameter narrowing).
_
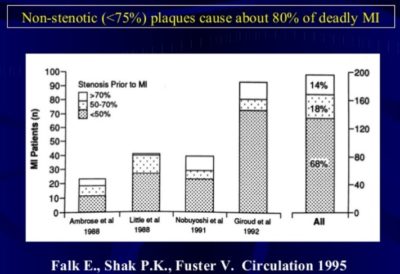
Plaque rupture is the most common type of plaque complication, accounting for ≈70% of fatal acute myocardial infarctions and/or sudden coronary deaths (Figure below). Several retrospective autopsy series and a few cross-sectional clinical studies have suggested that thrombotic coronary death and acute coronary syndromes are caused by the plaque features and associated factors. Most techniques for detecting and treating vulnerable plaque are devoted to rupture-prone plaque. This type of plaque has been termed a “thin-cap fibroatheroma.”
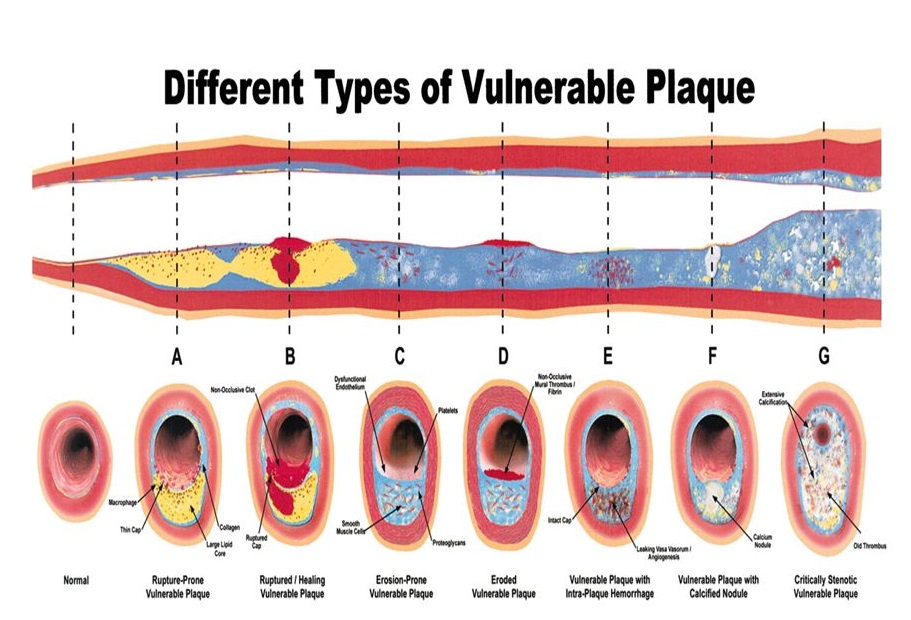
Figure above shows different types of vulnerable plaque as underlying cause of acute coronary events (ACS) and sudden cardiac death (SCD). A. Rupture-prone plaque with large lipid core and thin fibrous cap infiltrated by macrophages. B. Ruptured plaque with subocclusive thrombus and early organization. C. Erosion-prone plaque with proteoglycan matrix in a smooth muscle cell-rich plaque. D. Eroded plaque with subocclusive thrombus. E. Intraplaque hemorrhage secondary to leaking vasa vasorum. F. Calcific nodule protruding into the vessel lumen. G. Chronically stenotic plaque with severe calcification, old thrombus, and eccentric lumen.
_
Underlying Pathologies of “Culprit” Coronary Lesions:
Ruptured plaques (≈70%)
-Stenotic (≈20%)
-Non-stenotic (≈50%)
Non-ruptured plaques (≈30%)
-Erosion
-Calcified nodule
-Others/Unknown
_
Figure below shows phase and lesion morphology of coronary atherosclerosis.
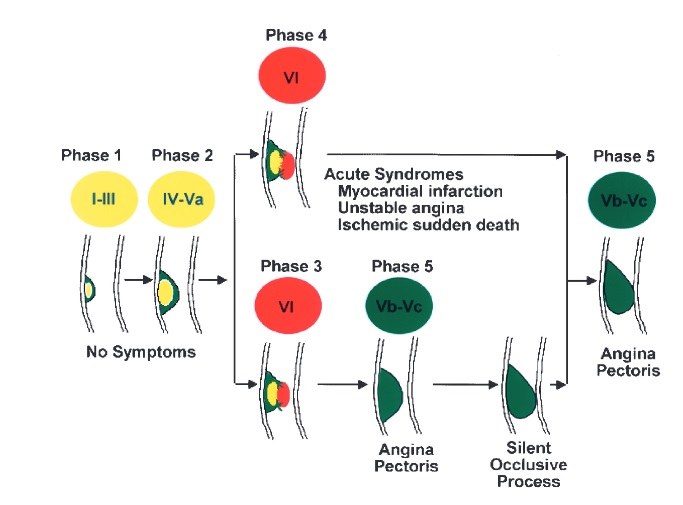
Progression is based on gross pathological and clinical findings. An early lesion (phase 1) can become a fibrolipid plaque (phase 2). Phase 2 can progress into an acute phase (phase 3 or 4). Formation of thrombosis or hematoma may cause angina pectoris (phase 3) or an ACS due to occlusive thrombosis (phase 4). Phase 3 and 4 lesions can evolve into a fibrotic phase (phase 5) characterized by more stenotic plaques that may progress to occlusive lesions. Yellow indicates lipid accumulation; red, thrombosis and hemorrhage; and green, fibrous tissue. Roman numerals indicate lesion types, as follows: I to III, early lesions with isolated macrophage-foam cells (I), multiple foam-cell layers (II), and isolated extracellular lipids (III); IV to Va, advanced lesions(fibrolipid plaques with confluent extracellular lipid pools [IV] and fibromuscular tissue layers and atheroma [Va]); VI, advanced lesions (complicated plaques with surface defects, hemorrhage, orthrombus deposition); and Vb to Vc, advanced lesions with calcifications (Vb) and those with fibrous tissue (Vc).
_
Plaque Composition in Acute Coronary Syndrome:
The term “acute coronary syndrome” summarizes various clinical presentations of acute myocardial ischemia and may be accompanied by a wide spectrum of clinical symptoms. It is classified into unstable angina, non–ST-segment elevation myocardial infarction, and ST-segment elevation myocardial infarction. Most ACS events are the result of sudden luminal thrombosis due to plaque rupture (two thirds) or erosion (one third). A ruptured plaque is characterized by a large necrotic core (usually > 30% plaque area in two thirds) and a luminal thrombus in continuity with the necrotic core through the gap in a thin (< 65 µm) ruptured fibrous cap. A plaque with a core shorter than 3 mm, with an area of less than 1.0 mm2, a percentage core of less than 10%, and a cap less than 150-µm thickness has a low likelihood of rupture. Compared with plaque ruptures, erosive plaques are characterized by fewer inflammatory cells and nonocclusive thrombi. Generally, the lipid core in ruptured plaque is larger than TCFA with intact fibrous cap (> 25% versus 10%). In ruptured plaques, necrotic cores are larger than 1.0 mm2 (3.8 ± 5.5 mm2) in 80%. In TCFA, the area of the necrotic core in at least 75% of cases is less than or equal to 3 mm2. However, the length of the necrotic core is similar in both, with a mean length of 8–9 mm (range, 2.0–22.5 mm). Overall, the mean cross-sectional area of narrowing is 70%, with 50% diameter stenosis. Plaque calcification is present in 69% of ruptures versus 23% of plaques with superficial erosions.
_
Not all vulnerable plaque ruptures, and researchers at the Texas Heart Institute are looking at ways to determine which vulnerable plaques are most likely to rupture. Some of our researchers are measuring the temperature of vulnerable plaque. They found that the warmer the plaque, the more likely it will crack or rupture. They are testing catheters that use infrared radiation and metal heat-sensing systems to measure the temperature of vulnerable plaque. Also, scientists at THI have discovered that vulnerable plaque has a low pH (is more acidic) and that such acidic plaques are more likely to rupture. The researchers are testing a device for the tip of a fiberoptic catheter that will allow them to measure the pH of plaque. Not all plaque ruptures lead to a heart attack or stroke. Studies have demonstrated that during a heart attack, several arteries are often involved with plaque rupture, yet not all develop occlusive thrombosis. Therefore, other factors are involved in determining the event severity and outcome.
_
Note:
Type-1 AMI is caused by an acute atherothrombotic coronary event; type-2 AMI is a more heterogeneous entity, where a condition other than coronary artery disease (CAD) contributes to an acute imbalance between oxygen supply (e.g., hypoxemia, anemia, hypotension) and demand (e.g., tachycardia, hypertension). In critically ill patients, or in patients undergoing major (non-cardiac) surgery, elevated values of cardiac biomarkers may appear, due to the direct toxic effects of endogenous or exogenous high circulating catecholamine levels. Also coronary vasospasm and/or endothelial dysfunction have the potential to cause type-2 AMI. Evidence-based treatment recommendations for type-1 AMI are clearly established, however for type-2 AMI these recommendations are lacking. Discussion on vulnerable plague is related to type-1 AMI and not type-2 AMI.
______
______
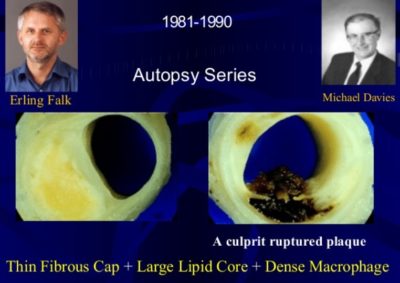
History of vulnerable plaque:
Today’s concept of vulnerable plaque has evolved primarily from the early pioneering work uncovering the pivotal role of plaque rupture and coronary thrombosis as the major cause of acute myocardial infarction and sudden cardiac death. Since the first historical description of plaque rupture in 1844, several key studies by leading researchers and clinicians have led to the current accepted views on lesion instability. Important to the complex paradigm of plaque destabilization and thrombosis are many discoveries beginning with the earliest descriptions of advanced plaques, reminiscent of abscesses encapsulated by fibrous tissue capable of rupture. It was not until the late 1980s that studies of remodeling provided keen insight into the growth of advanced plaques, beyond the simple accumulation of lipid. The emphasis in the next decade, however, was on a focused shift toward the mechanisms of lesion vulnerability based on the contribution of tissue proteolysis by matrix metalloproteinases as an essential factor responsible for thinning and rupture of the fibrous cap. In an attempt to unify the understanding of what constitutes a vulnerable plaque, morphological studies, mostly from autopsy, suggest the importance of necrotic core size, inflammation, and fibrous cap thickness. Definitive proof of the vulnerable plaque, however, remains elusive because animal or human data supporting a cause-and-effect relationship are lacking. Although emerging imagining technologies involving optical coherence tomography, high-resolution MRI, molecular biomarkers, and other techniques have far surpassed the limits of the early days of angiography, advancing the field will require establishing relevant translational animal models that produce vulnerable plaques at risk for rupture and further testing of these modalities in large prospective clinical trials. (Arterioscler Thromb Vasc Biol. 2010;30:1282-1292.)
_
Salute to Pioneers:
_
_
_
_
_
_
_

_______
_______
Vulnerable plaque Overview:
The concept of vulnerable plaque was first described by Dr. James E. Muller over 20 years ago, based on observations in studies of patients with acute myocardial infarction (MI), in whom he noted that these acute vascular events tended to occur most commonly early in the morning. He hypothesized that these event clusters must represent patients with “vulnerable plaques” that had become disrupted in response to some circadian early morning trigger. The trigger induced an abrupt transition from a lesion that was stable but vulnerable, to a frankly disrupted, thrombotic and clinically unstable culprit. His seminal work serves as the basis for our present concept that unstable plaques develop in “vulnerable patients” i.e. those who are at greatest risk of developing vulnerable lesions. A variety of technologies, both invasive and non-invasive, have been developed to try to characterize vulnerable plaques. Extensive work has also been done to delineate clinical characteristics that identify “vulnerable patients.” Abundant pathological and intravascular ultrasound (IVUS) data is now available to support the existence of one or more distinct plaque entities that fulfill the criteria of a “vulnerable plaque.” Based on these studies, TCFAs appear to be at least one common precursor to disrupted plaque. As a result, a variety of technologies, both invasive and non-invasive, have been developed to try to detect TCFAs in patients before they actually rupture and become clinically unstable. An appreciation of the information necessary to precisely characterize plaques is essential. The ideal tool would provide a complete roadmap of atherosclerotic burden throughout the coronary tree and provide data characterizing the architecture, composition and dynamic biology of each lesion. Comprehensive plaque analysis should optimally include the following parameters: 1) Architecture: plaque volume, length, eccentricity, remodeling and impact on lumen area; 2) Physiology: impact on coronary flow reserve; 3) Composition: lipid, fibrous tissue, calcium, etc; and 4) Patho-biology: presence of inflammation, neovascularization, fibrous cap metabolism, apoptosis, etc. Presently available and emerging technologies to detect vulnerable plaque include traditional grayscale IVUS, IVUS Virtual Histology, optical coherence tomography (OCT) and near infra-red (NIR) spectroscopy. These devices provide either structural or biochemical data that help characterize the architecture and biologic activity of atherosclerotic plaque. Each technology has strengths and limitations.
_
Vulnerable Plaque: Issue of Nomenclature:
The term ‘vulnerable’ plaque was coined by Muller and colleagues, to describe a plaque that by becoming disrupted has a high likelihood of starting the adverse cascade. There is disagreement over the meaning of this term, and several terms like high risk plaque, culprit plaque and unstable plaque have been used interchangeably to indicate the same pathological lesion.
The term ‘unstable plaque’ basically connotes an unstable clinical situation. It should therefore, be used only when vulnerable plaque has already initiated the clinical cascade of ACS. Because the term also has well-accepted clinical usage to describe unstable angina pectoris, confusion between the clinical syndrome and plaque is inevitable. Therefore, it is proposed that the term ‘unstable’ be reserved for the clinical syndrome and not for the plaque.
The terminology to describe vulnerability has become relatively standardized. The term “vulnerable plaque” is used to designate a plaque at high risk of disruption leading to thrombosis. Although there is general consensus regarding this terminology, existence of multiplicity of terms to describe the concept of vulnerability is confusing at times. But they also reflect different pathophysiological aspects of the phenomenon, hence enhancing our knowledge in this respect. Since the 1970s, scientists have been seeking the mechanisms responsible for converting chronic coronary atherosclerosis to acute coronary artery disease. As insights into this process have evolved, the relevant terminology has been continually updated. “High-risk plaque” and “thrombosis-prone plaque” are used as synonyms for “vulnerable plaque”. But it is encouraged to use the term “vulnerable plaque” for simplicity and uniformity.
Following consensus has emerged regarding the concept of vulnerability.
- Rupture-prone plaques are not the only vulnerable plaques. All types of atherosclerotic plaques with high likelihood of thrombotic complications and rapid progression should be considered as vulnerable plaques.
- Vulnerable plaques are not the only culprit factors for the development of acute coronary syndromes, myocardial infarction, and sudden cardiac death. Vulnerable blood (prone to thrombosis) and vulnerable myocardium (prone to fatal arrhythmia) play an important role in the outcome. Therefore, the term “vulnerable patient” may be more appropriate and is proposed now for the identification of subjects with high likelihood of developing cardiac events in the near future.
- A quantitative method for cumulative risk assessment of vulnerable patients’ needs to be developed that may include variables based on plaque, blood, and myocardial vulnerability.
_
Vulnerable Plaque Conundrum:
| What it originally meant |
| A plaque that is the culprit lesion for an acute coronary event |
| What it should mean |
| A plaque that is prone to rupture when all intrinsic and extrinsic effects are taken into account (regardless of structure) |
| How it is currently interpreted in the majority of literature |
| A plaque with specific morphological features—usually referring exclusively to thin cap fibroatheromas |
This table attempts to summarize the different connotations inherent in the term “vulnerable plaque.” It can be appreciated that a teleological approach has shifted toward a utilitarian one, allowing for easier classification of plaques as “vulnerable” or not, yet depriving this characterization of prognostic implications.
_
Vulnerable plaque versus culprit plaque:
Culprit plaque is a retrospective terminology. Whereas interventional cardiologists and cardiovascular pathologists retrospectively describe the plaque responsible for coronary occlusion and death as a culprit plaque, identification of such a plaque before any acute event, based on specific features is desirable and is the focus of intense research currently. Basic research and works based on pathological studies, invasive and non-invasive imaging have identified different morphological and biological features of atherosclerotic plaques which are prone to develop an acute coronary event. Many of these concepts revolve around identification of the plaques which are prone to rupture. But our understanding has evolved to recognize the fact that plaque rupture is but one of the mechanisms leading to thrombosis and acute plaque event. Existence of alternative mechanisms such as plaque erosion has come to be recognized. Prospective identification of such a plaque is central to the concept of “Vulnerable plaque”. Vulnerable plaque is thus, a future culprit plaque.
The term vulnerable plaque refers to all plaques at risk for thrombosis or rapid progression to become culprit lesions. A vulnerable plaque is not necessarily a soft plaque, a noncalcified plaque, an AHA type IV plaque, or a nonstenotic plaque.
_
Criteria for diagnosis of vulnerable plaque:
Naghavi et al have proposed diagnostic criteria based on histopathological features. Although based on retrospective studies, these criteria can be a useful reference point. Moreover, with availability of newer diagnostic tools many of the proposed features can be identified prospectively to make a correct diagnosis based on these.
Major Criteria:
- Active Inflammation (monocyte/macrophage infiltration): Plaques with active inflammation may be identified by extensive macrophage accumulation. Possible intravascular diagnostic techniques include thermography (measurement of plaque temperature), contrast-enhanced (CE) MRI, and fluorodeoxyglucose positron emission tomography. It has recently been shown that optical coherence tomography (OCT) reflects the macrophage content of the fibrous cap.
- A thin cap with a large lipid care: These plaques have a cap thickness of <100 µm and a lipid core accounting for >40% of the plaque’s total volume. Possible diagnostic techniques include OCT, intravascular ultrasonography (IVUS), MRI, angioscopy, near infrared (NIR) spectroscopy and radiofrequency IVUS analysis.
- Endothelial Denudation with Superficial Platelet Aggregation: These plaques are characterized by superficial erosion and platelet aggregation or fibrin deposition. Possible intravascular diagnostic techniques include angioscopy with dye and OCT. Noninvasive options include platelet/fibrin-targeted single photon emission computed tomography and MRI.
- Fissured/Injured Plaque: Plaque with a fissured cap that did not result in occlusive thrombi may be prone to subsequent thrombosis. Possible diagnostic techniques include OCT, IVUS, angioscopy and MRI. Fissured coronary plaques can be found in up to 25% of patients with CAD who died of noncardiac causes.
- Severe Stenosis: On the surface of plaques with severe stenosis, shear stress imposes a significant risk of thrombosis and sudden occlusion. The current standard technique is resonance angiography. Noninvasive options include multislice CT and magnetic resonance angiography with or without a contrast agent.
Minor Criteria:
- Superficial Calcified Nodules: These plaques have a calcified nodule within or very close to their cap and this structure protrudes through and can rupture the cap. This event may or may not be associated with severe coronary calcification and a high calcium score.
- Yellow Colour (on Angioscopy): Yellow plaques, particularly glistening ones may indicate a large lipid core and thin fibrous cap, suggesting a high risk of rupture. However, because plaques in different stages can be yellow and because not all lipid-laden plaques are destined to rupture or undergo thrombosis, this criterion may lack sufficient specificity.
- Intraplaque Hemorrhage: Extravasation of red blood cells or iron accumulation in plaque may represent plaque instability. Possible diagnostic technique includes NIR spectroscopy, tissue Doppler methods and MRI.
- Endothelial Dysfunction: Vulnerable plaques have sites of active inflammation and oxidative stress and are likely to be associated with impaired endothelial function. Possible diagnostic techniques are endothelium-dependent coronary artery dilatation and measurement of flow-mediated dilatation by brachial artery ultrasonography.
- Expansive (positive) Remodeling: Many of the nonstenotic lesions undergo “expansive,” “positive,” or “outward” remodeling i.e. compensatory enlargement before compromising significantly on the vascular lumen. As the luminal area was not affected, this phenomenon was considered as positive remodeling. Several studies have suggested that such remodeling is a potential surrogate marker of plaque vulnerability. IVUS was used in these studies to evaluate remodeling in coronary arteries.
_____
The concept of vulnerable plaque:
The concept of vulnerable plaque is based on the premise that certain coronary plaques are more prone to disruption or thrombosis than others, leading to a symptomatic acute coronary event. Histological investigations have revealed two main features of coronary plaques associated with acute thrombus formation, i.e. plaque rupture (in about 70% of cases) and plaque erosion (in about 30% of cases). Ruptured plaques are characterised by a large lipid pool of cholesterol-rich necrotic core covered by a thin layer of fibrous cap infiltrated with macrophages and T-lymphocytes. These plaques are supplied by an abundant vasa vasorum penetrating into the plaque intima from the adventitia. Conversely, the more infrequent plaque erosion is characterised by an absent or disrupted endothelial lining and greater proliferation of smooth muscle than inflammatory cells. Of note, plaque erosions often lack the large lipid pool and/or thin fibrous cap, and appear to be more common in younger women.
It is assumed that plaques vulnerable to rupture share the same morphological features as ruptured plaques, but with an intact thin fibrous cap of < 65 µm. These lesions are termed thin-cap fibroatheroma (TCFA) and are considered to be the requisite precondition for subsequent plaque rupture. Interestingly, a serial angiographic study showed that the actual size of plaques responsible for the infarction was only moderate, with the majority of lesions having less than 50% luminal narrowing before the event (which does not necessarily imply that high-grade stenoses are always “innocent” and unlikely to rupture, but are rather less frequently represented compared with mild lesions). Furthermore, an intravascular ultrasound (IVUS) study showed that rupture-prone plaques have large plaque burden expanding into the outer layer of the vessel wall, as reflected by positive remodeling. Thus, these plaques are often clinically silent (without inducing anginal symptoms or myocardial ischaemia) before the acute coronary event occurs.
To date, the multicenter Providing Regional Observations to Study Predictors of Events in the Coronary Tree (PROSPECT) trial was the first and largest natural-history study of atherosclerosis, using invasive angiography and IVUS among 697 patients with acute coronary syndrome (ACS) to identify plaques prone to rupture [vide infra]. In this study plaque burden ≥ 70%, minimal luminal area ≤ 4 mm2 and TCFA characteristics on VH were independently associated with subsequent major adverse cardiovascular events (MACE) derived from non-culprit lesions. For the first time it was demonstrated that the independent predictors of future events were the coronary plaque burden of ≥ 70%, the coronary minimal luminal area of ≤ 4.0 mm2, or the presence of TCFA. Of note, all of these characteristics were invisible to the coronary angiography but easily identifiable by IVUS. Attempts to identify and localise such vulnerable coronary plaques would be beneficial. A number of preventive measures, such as intensive lipid-lowering or antiplatelet treatment, as well as plaque sealing with coronary stents, could be implemented to effectively reduce the risk of future coronary events. To this end, there is a particular interest in developing a reliable non-invasive imaging method for the detection of vulnerable plaque, and CCTA is one of the leading candidates.
Markers of vulnerability at the plaque level include morphology such as cap thickness, the size of lipid core size, the colour of the plaque, collagen content, lipid content, stiffness, calcification and flow pattern throughout the coronary artery.
_
Table below shows Concepts related to vulnerable coronary plaque.
| Culprit lesion | Coronary lesion considered to be responsible for the clinical event, usually plaque complicated by intraluminal thrombosis |
| Thrombosed plaque | Plaque with an overlying thrombus extending into the vessel lumen either occlusive or non-occlusive |
| Eroded plaque | Thrombosed plaque (mainly fibrotic or proteoglycan-rich) due to loss or dysfunction of endothelial cells without associated rupture |
| Plaque with calcified nodule | Heavily calcified protruding plaque with loss or dysfunction of endothelial cells |
| Vulnerable, high-risk or thrombosis prone plaque | Plaque at increased risk of thrombosis and rapid stenosis progression TCFA: Inflamed plaque with a thin cap covering a lipid-rich necrotic core |
| Vulnerable patient | Patient at high-risk to experience a cardiovascular ischemic event due to a high atherosclerotic burden, high-risk plaques and/or thrombogenic blood and/or arrhythmogenic myocardium |
TCFA: Thin-cap fibroatheroma.
_____

The term vulnerable plaque was introduced by Muller et al in the late 1980s as a coronary plaque with a high susceptibility to rupture. Nowadays, the vulnerable plaque is commonly referred to as a lesion with a high likelihood of precipitating thrombosis. Recently, Stone et al introduced an alternative, more clinically relevant definition of the vulnerable plaque, that is, a plaque that places a patient at risk for future major adverse cardiac events (MACE). However, historically most pathophysiological studies on plaque morphology predisposing to ACS have focused on plaque rupture. A plaque with a large necrotic core and a thin fibrous cap is suspected to be rupture prone and is frequently referred to as a thin-cap fibroatheroma (TCFA).
_
Pathological Paradigm of the Vulnerable Plaque:
Medical term “vulnerable plaque” was first used by Muller et al. in defining the plaque with rupture that is one of the triggers for the onset of acute cardiovascular disease. Post-mortem studies of sudden cardiac death victims have shown that the major pathological substrates of coronary thrombosis are plaque rupture in 60–70 % of cases, plaque erosion in 20–30 % and thrombosis triggered by calcified nodules in 2–5 %. Plaque rupture, the most frequent mechanism of coronary thrombosis, involves disruption of a thin fibrous cap that overlies a large necrotic core, causing the thrombogenic contents of the necrotic core to come into contact with the bloodstream and trigger thrombus formation. Plaque erosion is defined as an abrasion of the endothelium without plaque rupture triggering thrombosis. Calcified nodules is a less common cause of coronary thrombosis, characterised by thrombus formation over nodular calcification protruding into the lumen through a disrupted thin fibrous cap. In current clinical practice, these non-rupture-type features in baseline lesions are also considered to be characteristics of vulnerable coronary plaques. In clinical setting, OCT is able to visualize plaque morphology possibly agree with autopsy findings. Furthermore, recent studies have shown that vulnerable plaques can develop not only in native coronary arteries but also in the neointima after long-term coronary stent implantation.
Pathological features of vulnerable plaque:
- Macroscopic features:
Large necrotic or lipid core
Luminal thrombus
Plaque hemorrhage
Spotty calcification
Positive vascular remodeling
- Microscopic features:
Thin fibrous cap covering lipid core
Macrophage infiltration
Plaque neovascularization
Endothelial denudation
Smooth muscle cell apoptosis
Protease expression
Endothelial adhesion molecule expression
- Heat generation
_
Figure below depicts a schematic representation of a vulnerable plaque, including the morphological aspects associated with rupture.
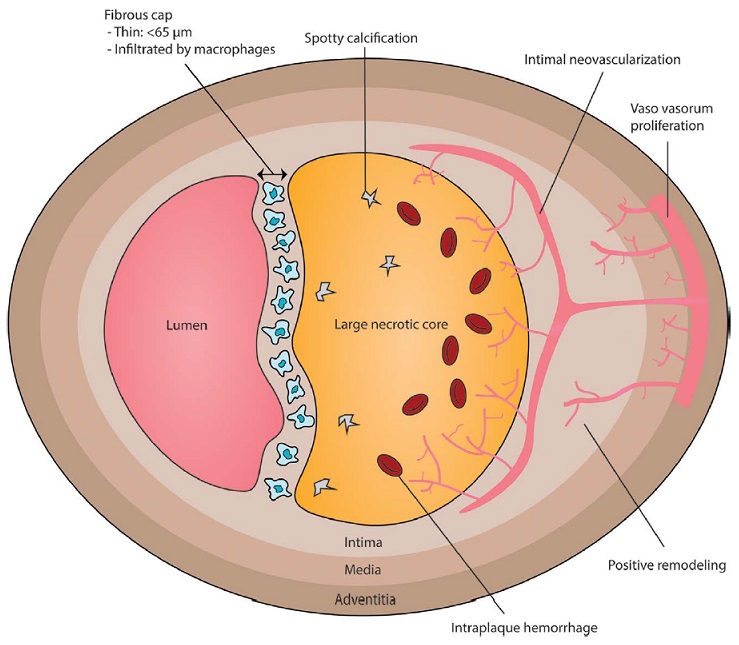
Figure above shows large necrotic core separated from the lumen by a thin fibrous cap (<65 µm) which is infiltrated by macrophages. The proliferation of the vaso vasorum leads to intimal neovascularization. These immature neovessels tend to leak red blood cells and cause intraplaque bleeding. There is positive (outward) remodeling of the vessel and the necrotic core contains spotty calcification.
_____
Vulnerable Plaque Morphology:
Autopsy studies have shown an inverse relationship between plaque cap thickness and risk of plaque rupture, with 95% of plaque ruptures occurring in lesions with a cap thickness of <65 µm. A postmortem study by Narula et al on 295 coronary atherosclerotic plaques analyzed the importance of various pathological characteristics to differentiate between TCFA and stable plaques and found that fibrous cap thickness was the most accurate discriminator. A large plaque burden with central necrosis negatively affects plaque stability. Expansion of the necrotic core may lead to erosion and subsequent thinning of the fibrous cap. Indeed, autopsy studies have revealed that ruptured plaques are associated with a larger necrotic core when compared with nonruptured plaques. Ruptured caps are more frequently infiltrated by macrophage foam cells than caps of nonruptured TCFA. It is hypothesized that macrophages within the cap play a key role in thinning and, ultimately, cap rupture by secreting proteolytic enzymes such as matrix metalloproteinases.
Coronary arteries may respond to plaque growth by expansion or shrinkage of the vessel wall, defined as positive and negative remodeling, respectively. Whereas vulnerable coronary plaques are generally associated with positive remodeling, stable plaques are characterized by negative remodeling of the vessel wall. Although positive remodeling may spare the coronary lumen, continuous outward growth of the plaque will induce neovascularization by microvessels originating from the adventitial vasa vasorum. Unfortunately, these microvessels are suspected to negatively affect plaque stability by causing intraplaque hemorrhage as a result of vascular immaturity and lack of supporting cells.
Plaque calcification is associated with all stages of atherosclerosis and can occur as early as during the intimal thickening phase. Microcalcification is thought to be triggered by apoptosis of smooth muscle cells in pathological intimal thickening lesions and by macrophage release of matrix vesicles or by apoptosis within the necrotic core in fibroatheroma lesions. Microcalcification within the thin fibrous cap, or spotty calcification, may serve as a marker of plaque vulnerability. In contrast, larger calcified sheets consisting of merged areas of microcalcification are associated with plaque stability. Whereas microcalcification within the thin fibrous cap and a specific calcification pattern called spotty calcification are reported to be associated with plaque rupture, predominantly calcified plaques with large calcific sheets are thought to be stable plaques.
_
Thin-cap fibroatheroma (TCFA) by definition comprises a large necrotic core covered by a thin fibrous cap of <65 μm. It is typically found in the proximal segments of coronary arteries with less than 50% diameter stenosis, and represents the most frequent type of lesion found in patients dying of coronary plaque rupture. By inference, when plaque composition is characteristic of a ruptured plaque, but there is an intact fibrous cap remaining, it is considered to predispose the “vulnerable plaque” to rupture. The natural course of coronary plaques with features of vulnerable plaque is not unidirectional, and it does not always involve a move toward plaque rupture manifesting as ACS, but rather it often remains asymptomatic, with ruptured plaques healing.
_
The morphological traits typically associated with rupture-prone plaques are found in lesions usually referred to as thin-cap fibroatheroma (TCFA) and are shown in figure below. They include a large eccentric necrotic core, occupying approximately one-quarter of the plaque area, a thin fibrous cap usually 65 μm, heavily infiltrated with macrophages and inflammatory cells, spotty calcifications, and vasa vasorum proliferation. Although two-thirds of acute events result from the rupture of a TCFA, the remaining events are caused by erosion of the intimal surface, with subsequent local thrombus formation. Eroded lesions usually exhibit less-severe luminal narrowing, fewer calcifications, less-extensive intraplaque neoangiogenesis, and less inflammation than ruptured plaques. By contrast, they usually demonstrate more negative than positive remodeling. These characteristics make them more difficult to detect than TCFAs.
_
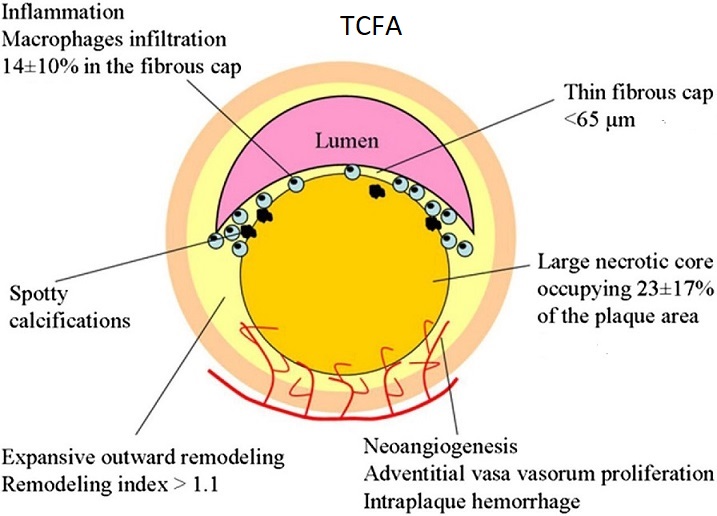
Figure above shows typical Morphological Traits associated With Rupture-Prone Plaques.
Thin cap fibroatheromas are most frequently observed in patients dying with acute myocardial infarction. They are most frequently observed in proximal coronary arteries, followed by mid and distal major coronary arteries. Vessels demonstrating TCFA do not usually show severe narrowing but show positive remodeling. In TCFAs the necrotic core length is approximately 2 to 17 mm (mean 8 mm) and the underlying cross-sectional area narrowing in over 75% of cases is <75% (diameter stenosis <50%). The area of the necrotic core in at least 75% of cases is ≤3 mm2. These lesions have lesser degree of calcification than plaque ruptures. Thin cap fibroatheromas are common in patients with high total cholesterol (TC) and high TC/high-density lipoprotein cholesterol ratio, in women >50 years, and in those patients with elevated high levels of high sensitivity C-reactive protein.
_
The current pathological definition of a vulnerable plaque is an inflamed thin fibrous cap overlying a necrotic-rich core. However, it is difficult to translate post mortem findings into a prospective, prognostically relevant concept for patients. The reported mean thickness of the fibrous cap of vulnerable plaques as assessed by pathologists differs as follows: Mann and Davies reported a mean cap thickness of 250μm (range 20–1,140μm) in type IV and V plaques, while Burke et al. reported a mean fibrous cap thickness of 23±19μm in ruptured plaques, and in 95% of these plaques cap thickness was <65μm. This cap thickness value for ruptured plaques has recently been widely used for the definition of non-ruptured TCFA. As atherosclerosis is a systemic disease, plaque rupture is considered a ubiquitous process that may be clinically silent or symptomatic in different regions of the vascular system. Chevuru et al. recently reported that the prevalence of TCFAs and ruptured plaques is low (0.46±0.95 and 0.38±0.70 per heart, respectively), focal and located in the proximal segments of the coronaries. The majority of TCFAs and ruptured plaques localised in the proximal third of the major coronary arteries, and in 92% of cases these lesions clustered within two or fewer non-overlapping 20mm segments. Necrotic core size was 1.6±1.8mm2 and 2.7±2.0mm long in TCFAs, while in ruptured plaques these measurements were 2.2±1.9mm2 and 1.9±3.6mm.
______
Vulnerability of the thin cap fibroatheroma.
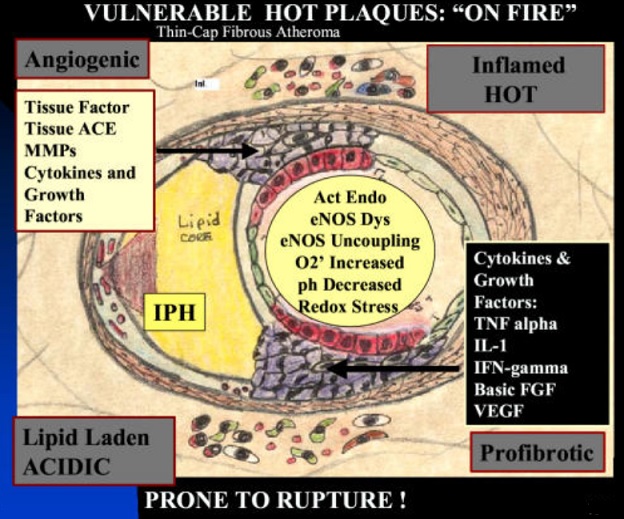
Figure above shows the vulnerable plaque: Currently the vulnerable plaque has been defined as containing the following: 1. Large lipid core. 2. Thin fibrous cap. 3. Inflammatory changes at the shoulder of the fibrous cap. 4. Decreased smooth muscle cells within the fibrous cap. This “Hot” – vulnerable thin-cap fibrous atheromatous plaque is associated with angiogenesis, inflammation, being lipid laden and acidic, and fibrotic. The endothelium is activated and these plaques are prone to rupture resulting in acute coronary syndromes.
______
| Markers of Vulnerability at the Plaque/Artery Level |
| MMP indicates matrix metalloproteinase; NO, nitric oxide; and IMT, intima medial thickness. |
| Plaque: |
| Morphology/Structure |
| • Plaque cap thickness |
| • Plaque lipid core size |
| • Plaque stenosis (luminal narrowing) |
| • Remodeling (expansive vs constrictive remodeling) |
| • Color (yellow, glistening yellow, red, etc) |
| • Collagen content versus lipid content, mechanical stability (stiffness and elasticity) |
| • Calcification burden and pattern (nodule vs scattered, superficial vs deep, etc) |
| • Shear stress (flow pattern throughout the coronary artery) |
| Activity/Function |
| • Plaque inflammation (macrophage density, rate of monocyte infiltration and density of activated T cell) |
| • Endothelial denudation or dysfunction (local NO production, anti-/procoagulation properties of the endothelium) |
| • Plaque oxidative stress |
| • Superficial platelet aggregation and fibrin deposition (residual mural thrombus) |
| • Rate of apoptosis (apoptosis protein markers, coronary microsatellite, etc) |
| • Angiogenesis, leaking vasa vasorum, and intraplaque haemorrhage |
| • Matrix-digesting enzyme activity in the cap (MMPs 2, 3, 9, etc) |
| • Certain microbial antigens (eg, HSP60, C. pneumoniae) |
| Pan-Arterial: |
| • Transcoronary gradient of serum markers of vulnerability |
| • Total coronary calcium burden |
| • Total coronary vasoreactivity (endothelial function) |
| • Total arterial burden of plaque including peripheral (e.g., carotid IMT) |
_____
What role does inflammation play?
Unstable plaque has a fibrous cap that “walls off” the lipid core of the plaque, which serves as a barrier, preventing the blood flowing through the lumen from meeting the thrombogenic lipid core. In patients with ACS, the fibrous cap becomes inflamed, thins and ruptures, exposing the clot-prone intraplaque “gruel” to clotting elements within luminal blood and thereby resulting in coronary thrombosis. Thus, inflammation appears to play a role locally at the point of plaque rupture. Inflammation appears to be a pan-coronary process, resulting in “multifocal” patterns of plaque instability and vulnerability. Angiographic observations demonstrate that nearly 50% of patients with acute MI manifest multiple sites of plaque instability. IVUS, angioscopic and pathological studies have confirmed that ACS patients harbor multiple sites of plaque rupture and, importantly, multiple TCFAs as well. It is important to note that inflammation exists not only locally within the coronary bed, but appears to be systemic. Patients with ACS typically show evidence of systemic inflammation, as indicated by elevated biomarkers such as C-reactive protein (CRP). Exactly what triggers inflammation in the systemic circulation and how it may impact the coronary circulation to induce plaque vulnerability and frank instability has been the subject of many hypotheses and intense scientific investigation.
Widespread vs. local inflammation:
Numerous clinical studies have shown that in patients who suffer an ACS, inflammation is not restricted only in culprit plaques. Widespread vascular inflammation in atherosclerosis is also supported by non-invasive imaging methods. In patients with peripheral artery disease, inflammation, as assessed by 18F-fluoro-D-glucose positron emission tomography-computed tomography (18FDG PET-CT scan), in paired arterial beds (carotid and femoral arteries) is highly correlated between left and right side. Similar conclusions have also been reached by our group using another non-invasive method for evaluation of carotid atherosclerotic plaque inflammation, microwave radiometry (MWR). Microwave radiometry allows in vivo non-invasive assessment of internal temperature of tissues, reflecting inflammation. Indeed, patients with multi-vessel coronary artery disease have higher carotid artery temperatures, compared with patients with one vessel or no coronary artery disease (CAD).
On the other hand, despite the widespread inflammatory background in atherosclerosis, culprit atherosclerotic plaques for cardiovascular events present with a higher degree of inflammation, underlining the importance of local factors in plaque destabilization. C-reactive protein has been detected in human coronary atherosclerotic plaques, suggesting its local production. Similarly, in patients with recent ischaemic stroke, culprit carotid arteries exhibit higher local temperature differences, compared with the contralateral carotids.
_______
Do we know if plaques like TCFAs are an early version of what later becomes hard, calcified plaque — or are there simply different types of plaque?
Atherosclerosis is a chronic disease punctuated by acute flares, some of which are clinically manifest as ACS, others of which occur “silently.” Thus, atherosclerosis progresses in an indolent pattern with some staccato bumps over decades. Plaques have various chemical and cellular constituents. Earlier stage lesions tend to be lipid-laden, then, as they advance, may contain variable volumes of lipid, fibrous tissue and cells (inflammatory and smooth muscle). Plaques may have a thin fibrous cap or a thick fibrous cap. Calcification tends to be a later stage in the process, but there is wide variation in composition from plaque to plaque, even within a single patient, i.e. patients may have plaques that are fibro-calcific as well as lipid-rich. They may have plaques that are TCFAs, and they may have plaques that are inflamed and ruptured. There are also transitions in the life of a plaque, in which it becomes eroded or frankly disrupted over time; intra-plaque hemorrhage is also an important pathophysiological process that contributes to plaque progression, instability and possibly calcification. To simplify, the TCFA can be viewed as an earlier stage of the plaque, whereas the fibro-calcific lesion is a more chronic scarred-down spectrum of the process.
_______
Researchers now think that vulnerable plaque is formed in the following way:
- Hyperlipidemia, hypertension, smoking, homocysteine, hemodynamic factors, toxins, viruses, and/or immune reactions results in chronic endothelial injury, dysfunction, and increased permeability.
- Lipoprotein LDL particles within the water/plasma portion of the blood stream, are absorbed into the intima, past the endothelium lining, some of the LDL-lipoprotein particles become oxidized and this attracts macrophages that uptake the particles. This process typically starts in childhood. To be specific: oxidized lipoprotein particles in the artery wall are an irritant which causes the release of proteins (called cytokines) which attract monocytes.
- The cytokines induce the endothelial cells lining the artery wall to display adhesion molecules that attract immune-system white blood cells (to be specific, monocytes).
- The monocytes squeeze into the artery wall. Once inside, they transform into macrophages which will ingest the oxidized lipoprotein particles.
- The macrophages sometimes become so overloaded with oxidized lipoprotein particles, the cholesterol contained therein and membrane-laden that they are called foam cells. Some of these cells die in place, releasing their fat and cholesterol-laden membranes into the intercellular space. This attracts more macrophages and smooth muscle cell migration and proliferation.
- Smooth muscle cells migrate from the media to the intima, proliferate, and develop an extracellular matrix made up of collagen and proteoglycans.
- In some regions of increased macrophage activity, macrophage-induced-enzymes erode away the fibrous membrane beneath the endothelium so that the cover separating the plaque from blood flow in the lumen becomes thin and fragile.
- Mechanical stretching and contraction of the artery, with each heartbeat, i.e. the pulse, results in rupture of the thin covering membrane, spewing clot-promoting plaque contents into the blood stream.
- The clotting system reacts and forms clots both on the particles shed into the blood stream and locally over the rupture. The clot, if large enough, can block all blood flow. Since all the blood, within seconds, passes through 5-micrometre capillaries, any particles much larger than 5 micrometers block blood flow. (most of the occlusions are too small to see by angiography).
- Most ruptures and clotting events are too small to produce symptoms, though they still produce heart muscle damage, a slow progressive process resulting in ischemic heart disease, the most common basis for congestive heart failure.
- The clot organizes and contracts over time, leaving behind narrowing(s) called stenoses. These narrowing(s) are responsible for the symptoms of the disease and are identified, after the fact, by the changes seen on stress tests and angiography, and treated with bypass surgery and/or angioplasty, with or without stents.
When this inflammation is combined with other stresses, such as high blood pressure (increased mechanical stretching and contraction of the arteries with each heart beat), it can cause the thin covering over the plaque to split, spilling the contents of the vulnerable plaque into the blood stream. Recent studies have shown cholesterol crystals within the plaque play a key role in splitting the plaque and also inducing inflammation. The sticky cytokines on the artery wall capture blood cells (mainly platelets) that accumulate at the site of injury. When these cells clump together, they form a thrombus, sometimes large enough to block the artery. The most frequent cause of a cardiac event following rupture of a vulnerable plaque is blood clotting on top of the site of the ruptured plaque that blocks the lumen of the artery, thereby stopping blood flow to the tissues the artery supplies.
Upon rupture, atheroma tissue debris may spill into the blood stream; this debris has cholesterol crystals and other material which is often too large (over 5 micrometers) to pass on through the capillaries downstream. In this, the usual situation, the debris obstruct smaller downstream branches of the artery resulting in temporary to permanent end artery/capillary closure with loss of blood supply to, and death of the previously supplied tissues. A severe case of this can be seen during angioplasty in the slow clearance of injected contrast down the artery lumen. This situation is often termed non-reflow. In addition, atheroma rupture may allow bleeding from the lumen into the inner tissue of the atheroma, making the atheroma size suddenly increase and protrude into the lumen of the artery, producing lumen narrowing or even total obstruction.
_
For many patients the first symptom of a ruptured vulnerable plaque is sudden coronary death. Yet not all sudden deaths can be tied to vulnerable plaques. If the plaque is in the main coronary vessel and it ruptures, your entire left heart is in jeopardy. If the plaque is in one of the small tributaries, you may suffer a heart attack. Vulnerable plaques are unique in that circulatory problems are not caused by the plaque’s occlusion of a vessel, but by the clot that is formed after the plaque ruptures. The destructive action of cholesterol on a vessel’s protective lining triggers the normal retaliatory action of monocytes, which “eat up” the molecules of cholesterol. This, in turn, draws more monocytes to the same area and, over time, a plaque forms. But because this plaque has a lipid core that extends into the vessel wall and is covered only by a thin, fibrous cap, it can erupt like a mini-volcano. And that sends cytokines (immunoregulatory proteins) into the bloodstream that attract more blood cells to form a thrombus. What actually causes these plaques to erupt or their fibrous caps to erode is still a mystery, although inflammation of the lesion is a prime suspect. Likewise, how “vulnerable” these plaques are to rupturing keeps cardiologists guessing. Until recently, vulnerable plaques have mostly been discovered during postmortem examinations. But new advances in imaging technologies are making it easier to discover them in living patients. And studies are now under way to identify those individuals who are at greatest risk.
_____
Detection:
While a single ruptured plaque can be identified during autopsy as the cause of a coronary event, there is currently no way to identify a culprit lesion before it ruptures. Several invasive and non-invasive strategies have been developed to assess the burden of vulnerable plaques. Intravascular ultrasound provides a two-dimensional cross-sectional image of the arterial wall and can help assess the plaque burden and composition. Optical coherent tomography offers superior resolution over intravascular ultrasound. High-resolution magnetic resonance imaging provides non-invasive imaging for visualizing fibrous cap thickness and rupture in plaques. In addition, it may be of value in assessing the effects of treatments, such as lipid-lowering therapy. Technical issues however limit its clinical applicability. Fractional flow reserve (FFR) may provide physiological functional assessment of plaque vulnerability; however, its role in the management of vulnerable plaque requires further studies.
Because artery walls typically enlarge in response to enlarging plaques, these plaques do not usually produce much stenosis of the artery lumen. Therefore, they are not detected by cardiac stress tests or angiography, the tests most commonly performed clinically with the goal of predicting susceptibility to future heart attack. In contrast to conventional angiography, cardiac CT angiography does enable visualization of the vessel wall as well as plaque composition. Some of the CT derived plaque characteristics can help predict for acute coronary syndrome. In addition, because these lesions do not produce significant stenoses, they are typically not considered “critical” and/or interventionable by interventional cardiologists, even though research indicates that they are the more important lesions for producing heart attacks.
The tests most commonly performed clinically with the goal of testing susceptibility to future heart attack include several medical research efforts, starting in the early to mid-1990s, using intravascular ultrasound (IVUS), thermography, near-infrared spectroscopy, careful clinical follow-up, and other methods, to predict these lesions and the individuals most prone to future heart attacks. These efforts remain largely research with no useful clinical methods to date. Furthermore, the usefulness of detecting individual vulnerable plaques by invasive methods has been questioned because many “vulnerable” plaques rupture without any associated symptoms and it remains unclear if the risk of invasive detection methods is outweighed by clinical benefit.
Another approach to detecting and understanding plaque behavior, used in research and by a few clinicians, is to use ultrasound to non-invasively measure wall thickness (usually abbreviated IMT i.e. intima-media thickness) in portions of larger arteries closest to the skin, such as the carotid or femoral arteries. Carotid IMT, especially those with >1-mm thickness, is now a well-recognized risk factor for atherosclerotic disease development in both carotid and coronary arteries.
______
Functional Versus Structural Assessment:
A growing body of evidence indicates that different types of vulnerable plaque with various histopathology and biology exist. To evaluate plaque vulnerability, it is evident that a combined approach capable of evaluating structural characteristics (morphology) as well as functional properties (activity) of plaque may be more informative and may provide higher predictive value than a single approach. For instance, a combination of IVUS or OCT with thermography may provide more diagnostic value than each of these techniques alone. Such an arrangement can be useful for both intravascular as well as noninvasive diagnostic methods. Autopsy and IVUS studies have shown that atherosclerotic lesions are frequently found in young and asymptomatic individuals. It is unclear what percentage of these lesions present morphologies of rupture-prone vulnerable plaques. Moreover, chronic inflammation and macrophage/foam cell formation are an intrinsic part of the natural history of atherosclerosis. These data suggest that screening only based on plaque morphology and/ or chronic markers of inflammation may not provide satisfactory predictive value for detection of vulnerable patients.
______
Does degree of luminal stenosis matter in Acute Coronary Syndrome?
A major finding in the last 2 decades was the recognition that plaque composition, rather than the severity of stenosis may determine the risk of thrombotic complications associated with ACS. It is now well established that disruption of the atherosclerosis lesion and the superimposed thrombus formation play a key role in the pathogenesis of ACS. A coronary artery study used angiography to prospectively evaluate nearly 3000 non-bypassed coronary segments in approximately 300 patients observed over a period of 42 to 66 months. It was shown that less obstructive plaques (< 80% stenotic at baseline) gave rise to more occlusion than severely obstructive plaques because of their greater number. Ambrose et al and Little et al first demonstrated that in approximately 50% of the cases of myocardial infarction, the lesions leading to occlusion were < 50% stenotic. Other groups have shown similar results, and today it is accepted that in approximately 70% of cases, the clot responsible for an acute coronary event occurs in a plaque that is < 50% stenotic. Therefore the assumption that only highly stenotic sites seen on angiography are at risk for thrombotic occlusion and subsequent myocardial infarction, whereas those coronary arteries that do not contain obstructive stenosis (< 50%) are nearly free of risk for thrombotic occlusion is not valid. It is likely that there are other factors that make a non-critically stenotic plaque unstable and prone to rupture.
_
There is increasing evidence that the degree of luminal stenosis alone insufficiently predicts the risk of subsequent plaque rupture in various arterial beds. It has been shown that the majority of myocardial infarcts (MI) occur in vessels with less than 50% stenosis. In a large, prospective natural history study of coronary atherosclerosis, Stone et al. recently demonstrated that lesions responsible for unanticipated events during the follow-up period were frequently angiographically mild (mean degree of stenosis 32%), most were thin-cap fibroatheromas (TCFAs; as assessed by intravascular ultrasound [IVUS]) or were characterized by a large plaque burden, a small luminal area, or some combination of these characteristics.

Figure above shows correlation between percentage of stenosis, frequency of plaques and risk of complication per plaque as a function of plaque progression. Although the average absolute risk of severely stenotic plaques may be higher than the average absolute risk of mildly stenotic plaques, there are more plaques with mild stenoses than plaques with severe stenoses.
Despite our advances in understanding the mechanisms of atherosclerotic disease and our knowledge that the degree of luminal stenosis is insufficient to predict a plaque’s vulnerability, current imaging markers that are included in the guidelines for carotid and coronary interventions are based solely on the degree of luminal stenosis.
There are several reasons for this paradox:
▪ Until recently, imaging of the arterial wall was not feasible and the only available imaging marker used in the large clinical trials that validated carotid endarterectomy was the degree of luminal stenosis, as assessed by digital subtraction angiography;
▪ It remains a matter of debate which plaque features should be assessed and which imaging modality should be used to assess a plaque’s vulnerability;
▪ Only a limited number of prospective outcome studies with relatively small number of patients based on plaque characterization have been performed;
▪ Lack of a representative animal model of plaque rupture and ACS/sudden death.
_
Complex angiographic appearance:
Coronary angiography in patients with ACS often reveals a complex plaque, which is an eccentric-appearing lesion with overhanging edges and irregular borders, ulceration, impaired flow, and often thrombosis. Complex plaques are an indicator of plaque rupture and/or thrombus in patients with unstable symptoms and have been considered to be the “culprit lesion”. Another report showed the presence of multiple complex plaques in the majority of patients with ACS, they required bypass surgery during admission more often and, during the subsequent year, had a significantly higher incidence of ACS. However, some complex plaques that undergo remodelling may become smooth, whereas others may remain complex but eventually become functionally stable, as angiographically complex lesions can also be found in a substantial percentage of patients with stable coronary artery disease (CAD). Thus, outside of clinically unstable phases, neither complex stenoses nor severe flow-limiting stenoses, which are typical of chronic stable angina, represent plausible, isolated, morphological markers of instability.
_
Remodelling of plaques:
In the early phases of atherosclerosis development, when CAD is minimal, luminal size is not affected by plaque growth because of the expansion of the external elastic membrane (EEM) and enlargement of the vessel size; this represents “positive remodelling”. Further, as CAD becomes moderate there is no increase in the vessel size, but rather the plaque approaches the lumen which shrinks; this is “negative remodelling”. Positive remodelling, larger plaque areas, and echolucent plaques are associated with unstable angina, while negative remodelling and smaller plaque areas are associated with stable angina. Positive remodelling is also seen in acute myocardial infarction at the site of plaque rupture. These findings are consistent with other observations that inflammation, calcification, and medial thinning are primary determinants of positive remodelling, which appears to be a feature of plaque instability.
_______
Broadening the understanding of vulnerable plaque:
According to Bernard De Bruyne, MD, PhD, of the Cardiovascular Center in Aalst, Belgium, screening for vulnerable plaque is “premature, too costly, and unproven.” Nonetheless, he said, he believes in the concept of vulnerable plaque. People who are at high risk and not being protected by optimal medical treatment should be targeted for treatment of local plaque. But, he emphasized, we are still a long way from accurate identification of such plaque and even farther from identification to effective treatment.
To begin with, the definition of “vulnerable plaque” is very loose, De Bruyne said, and many aspects of the phenomenon are underappreciated. For example, many people believe that thin cap fibroatheroma is the only pathological substrate for acute thrombotic occlusion. But there are many other culprits, he observed. For example, erosion of an atheroma is responsible for up to 30% of cases of ACS in women and diabetics. Another misconception is that vulnerable plaque is mostly non-stenotic and hemodynamically insignificant, he said. But there is good reason to suspect that in many patients with AMI, the stenosis is not benign. In a recent study in which careful quantitative coronary angiography was performed on patients with ACS, the vast majority were found to have significant stenosis underneath their thrombus. It is important to realize that these lesions induce a gradient, said De Bruyne, and that over time this force can contribute to plaque fatigue and rupture. In addition, when the lesion encroaches on the lumen, an area of low shear stress develops around the lesion that promotes plaque destabilization. In short, De Bruyne said, there is “cross-talk” among the hemodynamic, rheological, and biological contributors to vulnerable plaque.
______
______
Thrombosis:
Thrombus occurrence is the obligatory signature of a complicated vulnerable plaque. The thrombus formation may occur from elements issued from the plaque itself and/or the interaction between blood and activated platelets and dysfunctional endothelium. Several plaque components may be either directly or indirectly thrombogenic. Macrophage foam cells overexpress tissue factor. Tissue factor is a coagulation protein (via interaction with factor VII) that is secreted by the endothelium and macrophages, and is present in the plaque and in inflammatory conditions. It plays an important role in the thrombotic process. T lymphocytes induce tissue factor from macrophages. T lymphocytes, via the CD40 ligand, activate the macrophages and more particularly secretion of the tissue factor. Tissue factor secretion is also increased by oxidised LDL via the endothelial cells and smooth muscle cells. In vitro, oxidised LDL increases plasminogen activator inhibitor (PAI)-1, and decreases protein C and tissue plasminogen activator (t-PA) in endothelial cells. Lipoprotein Lp(a) participates in the promotion of thrombosis by interfering with plasminogen and overexpressing PAI-1. Increased concentrations of PAI-1 and apo(a) have been found in atherectomy samples of patients presenting with acute coronary syndromes; interestingly there was a correlation between concentrations of PAI-1 and apo(a) and macrophage density.
Blood also plays a role in the occurrence of local thrombus formation. Platelet activation is prompted by collagen exposure from the subendothelium: this represents a simple substratum of acute thrombosis in the setting of plaque erosion. Inhibition of platelet activation is reduced in unstable plaque by reduction of prostacyclin. The nature of the plaque itself may also participate in the thrombus formation. CD44 and proteoglycans, namely versican and hyaluronan, were found in high concentrations in eroded plaques. It has been suggested that hyaluronan could be involved in the development of thrombosis. CD44 receptors mediate platelet adhesion to hyaluronan. Hyaluronan also increases fibrin formation. Together these elements may facilitate the occurrence of thrombus in eroded plaques.
_
Post-treatment arterial thrombosis:
We have learnt from the two leading causes of thrombosis—erosion and ruptured plaques—that cell death (smooth muscle or endothelial) is a critical step. More generally, lack of healing results in thrombosis, namely by lack of endothelial regeneration and/or endothelial dysfunction, and lack of fibrous cap repair by smooth muscle cells. Thus, strategies aimed at inhibiting the healing process, with the goal of preventing restenosis after angioplasty, carry the risk of favouring thrombosis. Indeed, brachytherapy and coated stents with molecules acting on the cell cycle have shown the occurrence of late thrombosis. The treated lesion exhibits an atherosclerotic plaque with neointimal necrosis, cell death in experimental models, and “black holes”—that is, loss of neointima behind the cell struts in humans detected by intracoronary ultrasound. Indeed, the healing process which routinely covers the struts of a bare stent is impaired by active coated stents targeted against cell proliferation, thereby inhibiting endothelial coating and extracellular matrix repair, whereas restenosis is inhibited.
_______
_______
Vulnerable plaque studies:
There is growing interest in the possibility that identification and treatment of vulnerable plaques and vulnerable patients can enhance the progress made against coronary artery disease. Innovations in medical therapy—statins and other agents—and novel interventional cardiology techniques—e.g., drug-eluting stents—have significantly decreased the morbidity and mortality caused by coronary atherosclerosis. However, coronary events continue to be the leading cause of death worldwide. Improved preventive measures are needed because, for many individuals, sudden coronary death is the first sign of the disorder. And even those who survive an acute coronary syndrome remain at high risk. After successful treatment of the initial culprit lesion by a percutaneous coronary intervention (PCI), the risk of a coronary event from a new lesion is ≈10% in the following year and 5% in each of the subsequent 4 years as seen in the figure below.
Figure above shows occurrence of coronary events (revascularization, death, MI, acute coronary syndromes, or congestive heart failure) after PCI in 4 studies of bare-metal stents. In addition to events resulting from stented lesions (solid line), many events are caused by nontarget (vulnerable) plaques (dashed line).
These substantial levels of ongoing morbidity and mortality have led to heightened interest in new methods to prevent coronary events. For primary prevention, the effort has focused on plasma markers and noninvasive testing to identify vulnerable individuals. For secondary prevention, interest has focused on vulnerable patients and the vulnerable plaques they may possess that might be identified and treated during the catheterization for their initial event.
______
Study paradigm of vulnerable plaque:
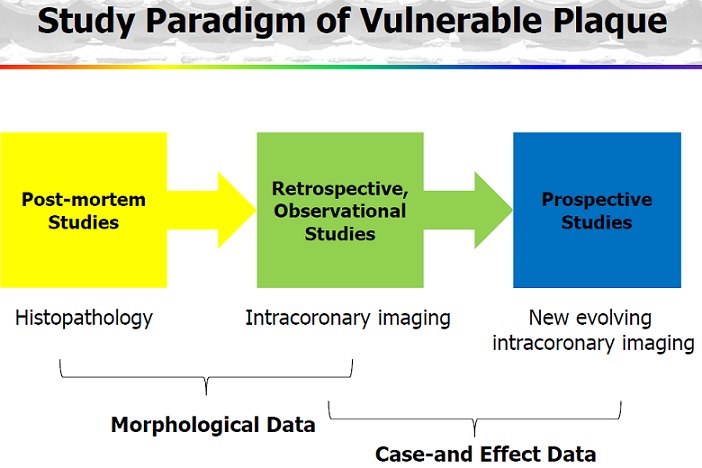
______
The extensive studies described below have great promise to enhance the detection and treatment of vulnerability and thereby prevent coronary events. The net result of improved risk stratification will be better assessment of the risk/benefit relationships for the novel therapies that are needed. This balance will require increased attention because novel antiatherosclerotic agents and treatments are likely to be costly and may carry unanticipated side effects.
As indicated in table below, many of the trials required to test the hypothesis that diagnosis and therapy of vulnerability are possible and beneficial are planned or in progress. In aggregate, the planned trials account for >6000 patients.
| Summary of Prospective Clinical Trials (in Progress or Planned) to Determine the Outcome Associated with Plaques Characterized by Intracoronary Diagnostic Devices | |||||
| Technique to Be Evaluated | Name of Trial | Trial Design | Types and Number of Patients to Be Enrolled | Principal Investigator | Sponsor |
| ACS indicates acute coronary syndromes. | |||||
| Shear stress profiling | Prediction | Changes in plaque morphology and remodeling at sites with low shear stress, plus major adverse cardiac events | 500 PCI | Dr Peter Stone | Boston Scientific, Inc |
| IVUS-based plaque composition | Prospect | Natural history for coronary events | 700 PCI | Dr Gregg Stone | Guidant, Inc, Volcano, Inc |
| IVUS-based plaque composition | Study of Prospective Events in Coronary Intermediate Atherosclerotic Lesions | Lesion progression and natural history for coronary events | 2000 PCI, including 1000 with repeat IVUS measurements at 12 months | Drs Etsuo Tsuchikane and Tadanori Aizawa | Volcano, Inc, Goodman Co, Fukuda Denshi Inc |
| Palpography | Substudy of Prospect | Natural history | 200 PCI | Dr Gregg Stone | Guidant, Inc |
| Palpography | IBIS-2 | Assessment of treatment with inhibitor of lipoprotein-associated phospholipase-2; End points are palpography and hs-CRP | 450 PCI | Dr Patrick Serruys | GlaxoSmithKline, Inc |
| Thermography | Vulnerability Index Program 1 | Feasibility and tolerability of thermal measurement | 160 PCI stable angina | Drs William Wijns and Stephan Verheye | Bristol Myers Squibb Medical Imaging, Inc |
| Thermography | Vulnerability Index Program 2 | Natural history for coronary events (after Vulnerability Index Program 1) | 700 PCI ACS | Drs William Wijns and Stephan Verheye | Bristol Myers Squibb Medical Imaging, Inc |
| NIR spectroscopy | Spectroscopic Assessment of Coronary Lipid plus Registry | Spectra in patients, followed by natural history for coronary events | 2000 PCI | Dr Sergio Waxman | InfraReDx, Inc |
| OCT | NIH-funded OCT Study | Stenosis progression on 18-mo restudy | 100 PCI | Drs Guillermo Tearney and Brett Bouma | NIH |
| Diagnostic technique to be selected | Pilot Safety Study for Prevail | Randomization of intermediate stenoses to stenting or medical therapy, with possible use of a vulnerability detector | 700 PCI | Dr Sheldon Goldberg | Cordis, Inc |
_
Although the efforts required to test the vulnerability hypothesis are considerable, they are justified by the benefits that could be achieved. Effective screening and treatment in the general population would decrease sudden death and initial MI, and success in the catheterization laboratory would decrease the high risk of recurrence that now follows PCI. Because the costly disorders of heart failure and arrhythmia would be prevented, economic analysis indicates that treatment of vulnerability would actually lower healthcare costs while decreasing morbidity and mortality resulting from coronary artery disease.
_
A common problem with associating plaque features and events is confounding. Increasing coronary atherosclerotic disease burden is strongly linked to greater event risk and the number of vessels with coronary artery stenoses (≥50%) correlate with plaque burden. Furthermore, the assignment of culprit lesions by lumen obstruction leads to stronger associations with features that are linked to stenoses. For example, baseline plaques with greater lumen obstruction are more likely to have larger plaque burden, calcification, low density plaque, etc., but they are also more likely than smaller lesions to develop into obstructive stenoses overtime (and then being labeled “culprit”). Therefore, many plaque characteristics may convey increased hazard of events merely through their association with a higher-grade stenosis. It is conceivable that some of the features lead to additive prediction of stenoses at follow up. For example, a baseline lesion with ≥50% lumen obstruction and large plaque burden is more likely to increase in stenosis severity than lesions with either characteristic alone. There is strong, consistent evidence that plaque ruptures occur frequently without causing events. Indeed, plaque ruptures appear to be part of the common pathway of plaque growth. It is only the exceptional case of plaque rupture or erosion that leads to an event, a perfect storm scenario. Given the very low absolute risk associated with individual plaques, the critical question is what intervention would be beneficial in this setting—even if identification of such plaques would be possible at low risk and costs. Any mechanical intervention by cardiac catheterization involves much greater risk than the risk of an acute coronary event associated with the lesion. It remains to be seen if specific medical treatment options, such as PCSK-9 inhibitors or anti-inflammatory drugs, can be justified in this scenario.
Coronary heart disease is a systemic illness which requires a comprehensive approach. We know that control of blood pressure, lipids, blood glucose, smoking cessation, exercise and diet have a profound effect on patient outcome. Unfortunately, data from clinical trials suggest we miss prevention goals in most patients. Achieving better prevention is likely to have a profound impact on the risk of adverse events. Assessing the total burden of atherosclerotic disease and its metabolic activity as well as considering systemic risk, such as inflammation, is more likely to aid in the management of patients with coronary heart disease than fixating on individual plaques. More than a decade after a major collaborative effort to move the field away from our focus on individual, “vulnerable” plaques, we still have not fully embraced the concept of the vulnerable patient.
_______
The Vulnerable Atherosclerotic Plaque: Scope of the Literature Free, a 2010 review:
The scope of recent literature on the concept of vulnerable plaque was reviewed by examining 463 abstracts of primary and review articles identified through MEDLINE (2003 to April 2010). Proposed definition criteria of vulnerable plaque included active inflammation, a thin cap with a large lipid core, endothelial denudation, fissured cap, severe stenosis, or combinations of these findings. In 242 primary studies, histopathology, biomarkers, and imaging of carotid and coronary artery plaques were evaluated for features suggestive of vulnerability. Notably, 89% of these studies were cross-sectional in design and were exclusively conducted in patients with known cardiovascular disease. None of the imaging studies documented whether the identified lesions were responsible for cardiovascular events. Cross-sectional design precludes evaluation of the predictive utility of biomarkers. Because vulnerable plaque is not an established medical diagnosis, no studies have been done that explicitly evaluate the treatment of vulnerable plaques. Few studies examined potential systemic treatments (for example, statins) to modify vulnerability features. Large prospective studies in patients with and without previous cardiovascular events during long follow-up are required to validate this concept.
Key Summary Points:
- The concept of the vulnerable plaque has been proposed as a paradigm to improve the prevention of cardiovascular disease by identifying atherosclerotic plaques at higher risk for causing future cardiovascular events.
- Proposed definition criteria of vulnerable plaque, based on examining plaques that have already caused a clinical event, include active inflammation, a thin cap with a large lipid core, endothelial denudation, fissured cap, severe stenosis, or combinations of these findings.
- Studies on biomarkers of plaque vulnerability are cross-sectional and thus cannot provide information on the predictive value of examined associations.
- Imaging studies are almost all limited by cross-sectional or retrospective design, diverse imaging methods or imaging features studied, and lack of reliable documentation on whether identified lesions were responsible for future cardiovascular events in the few prospective studies.
- Studies on therapies lack a standard definition of plaque vulnerability and are limited by the obligatory surrogate nature of the outcomes studied.
- Substantial conceptual and methodological challenges need to be addressed to realize the potential promise of the vulnerable plaque concept.
______
Number of ruptured plaques in addition to culprit lesion, a 2002 study:
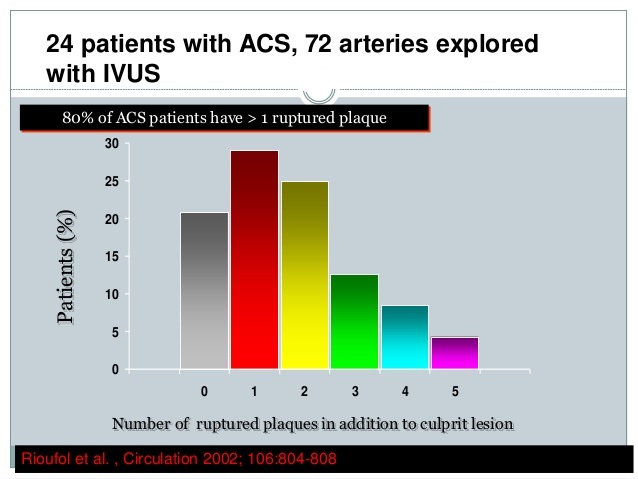
Figure above shows that 80% of ACS patients have > 1 ruptured plaque in this study.
______
PROSPECT: A Prospective Natural-History Study of Coronary Atherosclerosis:
Major adverse CV events after PCI in patients with ACS attributed to culprit, non-culprit lesions:
IVUS Assesses Risk of Future Vulnerable Plaque Ruptures, a 2011study:
Intravascular ultrasound (IVUS) enabled physicians to more accurately assess the risk of individual blockages than the use of the current standard of angiographic imaging alone, according to this new study. The results from the PROSPECT (Providing Regional Observations to Study Predictors of Events in the Coronary Tree) study were published in The New England Journal of Medicine. Investigators used grayscale IVUS and Volcano Corporation’s Virtual Histology (VH)-IVUS tissue characterization software to evaluate suspected vulnerable plaques in the coronary arteries. PROSPECT is the first study that is trying to identify vulnerable plaques. Until now, it’s been impossible to distinguish between soft, pliable plaques filled with lipids and hard, stable plaques. But this study goes beyond just identifying vulnerable plaque and it can predict which plaques and which patients are going to be in trouble. To better visualize plaque, researchers are using Virtual Histology Intravascular Ultrasound (VH IVUS) technology which gives a clearer picture of a plaque’s morphology and can even detect lipid in a plaque. VH IVUS uses advanced spectral analysis techniques and classifies atherosclerotic plaque color images into four component types: fibrous, fibro-fatty, dense calcium and necrotic core. It then uses a predetermined color key to display plaque compositions. Using this powerful imaging tool appears to have made a difference in the PROSPECT study.
The PROSPECT study is the first and only natural history study that tracked the clinical outcomes, hospital readmissions and subsequent coronary events of patients with acute coronary syndrome (ACS). In PROSPECT, 697 patients presenting with ACS underwent PCI followed by quantitative angiography of each millimeter of the entire coronary tree. In addition, the proximal 6 to 8 cm of all 3 major coronary arteries were imaged using IVUS. The investigators assessed these patients’ coronary arteries by taking very detailed measurements and interior images of all three arteries. The PROSPECT study showed that of the 55 imaging parameters measured, grayscale IVUS and VH-IVUS identified the only individual parameters that could statistically predict lesion risk for future clinical events. When combined together, grayscale IVUS and VH-IVUS uncovered lesion characteristics that PROSPECT showed predict the highest lesion risk for future clinical events. PROSPECT shows us that we can go beyond the limitations of a two-dimensional X-ray and better separate these blockages into higher risk and lower risk.
The investigators found that certain high-risk lesions, including those with a relatively small lumen area and a large amount of plaque and a significant amount of dead tissue or necrosis near the lumen as identified by VH-IVUS, had an almost 20 percent risk of a cardiac event within three years. These lesions were classified as “predictive” by the investigators. Conversely, certain low-risk lesions where that same necrotic tissue was not present in the study had a significantly lower event rate of only 0.6 percent out to three years. These lesions were termed “protective” by the investigators.
The landmark PROSPECT study has confirmed two key principles that will be relevant as we continue to innovate and improve our treatment of cardiovascular disease. First, angiographic guidance alone is not a good predictor of future events; grayscale intravascular ultrasound is much better than angiography at determining the severity of plaque, and which blockages are most likely to cause future events. Second, all coronary artery disease is not the same, and differences in underlying tissue type can in fact change the risk profile of a lesion. Some blockages are more active and can quickly progress to a clinical event. Other blockages are more stable, and when combined with proper medical therapy, have an extremely low risk of causing a future event. For the first time, we have been able to prospectively identify these lesions both by size and by tissue type using VH-IVUS.
PROSPECT: Conclusions:
- Approximately 20.4 % of pts with ACS successfully treated with stents and contemporary medical Rx develop MACE within 3 years, with adverse events equally attributable to recurrence at originally treated culprit lesions (treatment failure) and to previously untreated non-culprit coronary segments
- Most lesions causing non-culprit lesion events arose from angiographically mild (50%) stenoses; the mean angiographic diameter stenosis of the 106 non-culprit lesions subsequently responsible for MACE increased (from 32.3 at baseline to 65.4% at follow-up), often with new plaque rupture and/or thrombus.
- Significant independent predictors of non-culprit lesion related MACE were plaque burden >70%, presence of a TCFA by RF-IVUS and a minimal lumen area 4.0 mm2. The presence of 2 or 3 of these 3 characteristics identified lesions with a 10% and 18% likelihood, respectively, of an event arising from that site within 3 years. All of these characteristics were invisible to the coronary angiography but easily identifiable by IVUS.
- This prospective study therefore demonstrated that the plaques likely to cause future coronary events are indeed typically severe, either with a large plaque burden and/or a small minimal lumen area.
Final conclusion:
In patients who presented with an acute coronary syndrome and underwent percutaneous coronary intervention, major adverse cardiovascular events occurring during follow-up were equally attributable to recurrence at the site of culprit lesions and to non-culprit lesions. Although non-culprit lesions that were responsible for unanticipated events were frequently angiographically mild, most were thin-cap fibroatheromas or were characterized by a large plaque burden, a small luminal area, or some combination of these characteristics, as determined by gray-scale and radiofrequency intravascular ultrasonography.
______
Clinical and Angiographic Characteristics of Patients Likely to Have Vulnerable Plaques: Analysis From the PROSPECT Study, a 2013 analysis:
The present study demonstrated that clinical and angiographic characteristics had poor predictive accuracy in identifying patients with untreated high-risk plaques related to future adverse events. This finding highlights the potential value of comprehensive 3-vessel imaging assessment (either invasive or noninvasive) to evaluate plaque phenotype for more accurate risk stratification of patients admitted with ACS.
The PROSPECT, VIVA (VH-IVUS in Vulnerable Atherosclerosis), and PREDICTION (Prediction of Progression of Coronary Artery Disease and Clinical Outcome Using Vascular Profiling of Shear Stress and Wall Morphology) studies provided robust evidence that atheroma burden and the TCFA phenotype are strongly associated with plaque vulnerability. The present analysis for the first time showed that the coronary plaque phenotype, which is independently associated with non-culprit lesion–related MACE, cannot be predicted by the clinical and angiographic findings, emphasizing the potential value of more comprehensive plaque characterization (with either intravascular or noninvasive imaging) for more accurate risk stratification. Advances in intravascular imaging are expected to allow complete plaque characterization, and thus, an assessment of the whole coronary tree with upcoming invasive imaging techniques is anticipated to allow more accurate risk stratification. However, the additional time and cost, risk of complications, increased time required to process the obtained data, and lack of evidence about how we should treat high-risk patients make unrealistic the regular application of 3-vessel invasive imaging in clinical settings.
Noninvasive imaging carries a great potential because it overcomes the abovementioned limitations of intravascular imaging modalities. Computed tomographic coronary angiography appears able to provide information about the type of plaque, and there are robust data demonstrating its prognostic value in asymptomatic or symptomatic stable patients. Nevertheless, further research is required to examine its potential predictive ability in patients with acute myocardial infarction.
_______
_______
Vulnerable patient:
There are many factors that influence MI and strokes besides vulnerable plaque as seen in the figure below:
_
It is important to identify patients in whom disruption of a vulnerable plaque is likely to result in a clinical event. In these patients, other factors beyond plaque (i.e., thrombogenic blood and electrical instability of myocardium) are also responsible for the final outcome. In fact, plaques with similar characteristics may have different clinical presentations because of blood coagulability (vulnerable blood) or myocardial susceptibility to develop fatal arrhythmia (vulnerable myocardium). The latter may depend on a current or previous ischemic condition and/or a nonischemic electrophysiological abnormality.
Hence, the term “vulnerable patient” has been introduced to indicate an individual with a high likelihood of experiencing a cardiac event. Such a patient is likely to have vulnerable blood (prone to thrombosis), vulnerable myocardium (prone to arrhythmia), and ≥1 vulnerable plaque. To enhance primary prevention, a search is underway for novel methods to identify vulnerable patients. Because a vulnerable plaque is likely to be the root cause of vulnerability of the patient and may be amenable to systemic or local therapy, the search for the vulnerable patient of necessity is closely linked to efforts to identify vulnerable plaques.
_
Our current understanding is driven by the recognition that potentially life-threatening events are caused not only by vascular changes but also by alterations in the nature of the circulating blood and the myocardium. As such, these three components—vulnerable vessel, vulnerable blood, and vulnerable myocardium—characterize what is today referred to as the vulnerable patient, who is defined as a patient with a high risk for the development of cardiovascular complications as seen in the figure below:

Figure above shows that the risk of a vulnerable patient is affected by vulnerable plaque and/or vulnerable blood and/or vulnerable myocardium. A comprehensive assessment must consider all of the above.
The recognition that cardiovascular risk is determined not only by the vulnerable vessel but also by components of the vulnerable blood and the vulnerable myocardium has major implications for future research and therapeutic developments. To achieve a more precise and accurate risk stratification in the future, novel risk markers should be identified, taking into account all three components of the vulnerable patient.
_
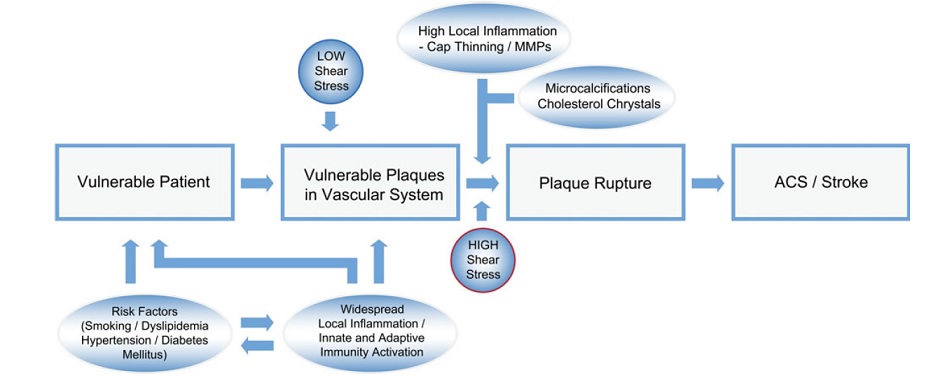
Figure above shows schematic illustration of pathobiological mechanisms implicated in plaque vulnerability and rupture. Vulnerable patient’s risk factors establish a prothrombotic microenvironment. There is a strong interplay between risk factors and widespread inflammation in the vascular tree. Hemodynamic factors (endothelial shear stress/wall stress) act locally on a vulnerable plaque, characterized by high inflammatory activation, cholesterol crystals, and microcalcifications, leading to plaque rupture and potentially to clinical events. ACS, acute coronary syndrome; MMPs, matrix metalloproteinases.
______
______
An update on new pathophysiological mechanisms implicated in vulnerable plaque formation and rupture:
Inflammatory activation in atherosclerosis includes both innate and adaptive immunity mechanisms. Macrophages, recruited as monocytes from the circulating blood, are the predominant cell type of innate immunity in atherosclerosis. The recruitment takes place through adhesion molecules (vascular cell adhesion molecule-1) expressed by vascular endothelial cells (ECs). In the plaque, these mononuclear phagocytes express scavenger receptors necessary for uptake of modified lipoproteins and, hence, foam cell formation. Macrophages of the plaque can overproduce matrix metalloproteinases (MMPs), specific enzymes with interstitial collagenase activity, causing the thinning of fibrous cap.
It is now well recognized that at least three subtypes of macrophages can be found in atherosclerotic lesions: M1, the most common form, is a pro-inflammatory macrophage subtype, activated by cytokines secreted by T cells, while M2 seems to play an anti-inflammatory role, promoting tissue repair. The activation of M1 macrophages is mediated by toll-like receptor-4 (TLR-4). A novel third atheroprotective macrophage phenotype, haemoglobinstimulated macrophage (M(Hb)), induced by intraplaque haemorrhage, has also been isolated in atheromas. Cumulative experimental and clinical evidence imply an important role of adaptive immunity in atherosclerosis progression and destabilization. Indeed, the cells of adaptive immunity, namely T and B lymphocytes, reside in atherosclerotic plaques.
_
MicroRNAs in vulnerable plaque formation and rupture:
MicroRNAs (miRNAs) are small, non-coding post-transcriptional regulatory RNAs. They inversely regulate their target gene expression at the post-transcriptional level either by inhibiting translation or by causing degradation of the target messenger RNA (mRNA). Their potential role in atherosclerotic plaque formation and rupture, through regulation of inflammation, microcalcification, angiogenesis and apoptosis, and interaction with biomechanical factors, has recently been the focus of extensive research. MicroRNAs seem to participate as post-transcriptional regulators in all stages of the inflamed atherosclerotic plaque formation, ending to its rupture. VCAM-1 expression by ECs, inflammatory activation of plaque macrophages in M1 phenotype, lipid accumulation in macrophages and foam cell formation, oxLDL-induced lipid uptake through regulation of scavenger receptors, and macrophage apoptosis are only few of the processes regulated.
_____
_____
Biomechanical factors in vulnerable plaque formation and rupture:
So far we discussed characteristic histomorphologic features of vulnerable plaques including a high lipid content, increased numbers of inflammatory cells, and extensive adventitial and intimal neovascularity as a prerequisite for plaque disruption. The fibrous cap of an atherosclerotic plaque may become thin and rupture as a result of the depletion of matrix components through the activation of enzymes, such as matrix-degrading proteinases and cysteine and aspartate proteases, and through the reduction in the number of smooth muscle cells. Activated T cells may also inhibit matrix synthesis through the production of interferon-gamma.
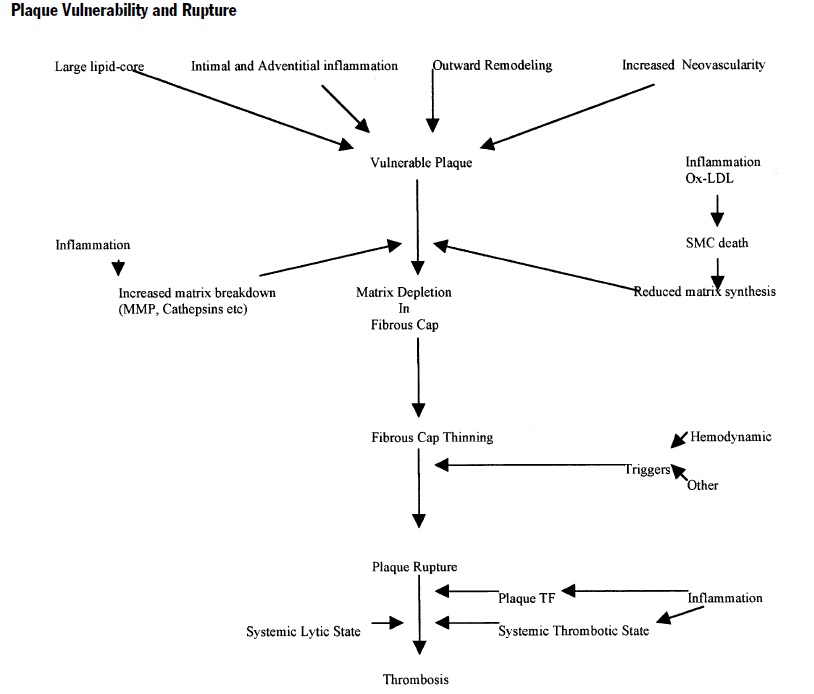
Figure above shows conceptual model depicting the potential pathophysiologic mechanisms of plaque vulnerability, rupture, and thrombosis. Ox-LDL = oxidized low-density lipoprotein; MMP = matrix degrading metalloproteinases; SMC = smooth muscle cells; TF = tissue factor.
_
Vulnerable plaque morphology has been described by gross pathology and intravascular ultrasound, but morphological criteria cannot fully explain vulnerability, which involves four distinct factors: 1) inflammatory and biological processes; 2) geometry; 3) composition; and 4) hemodynamic stress. These last three aspects underlie the biomechanical study of vulnerable plaque.
By virtue of the nature of their evolution, atherosclerotic plaques tend to be eccentric, and this is a crucial morphological feature, causing circumferential stress to peak in very specific juxta-luminal locations, where it can exceed the rupture threshold of collagen, the basic constituent of arterial architecture. The lipido-necrotic core covered by a fibrous cap, formed in young plaques, is another morphological feature, which, can also increase and concentrate circumference stress in the juxta-luminal fibrous cap. The larger the lipid core, the thinner the fibrous cap and the greater is the stress. There are also inflammatory processes in such areas, which tend to reduce cap thickness. Ruptures occur when this thickness falls below 65 microns.
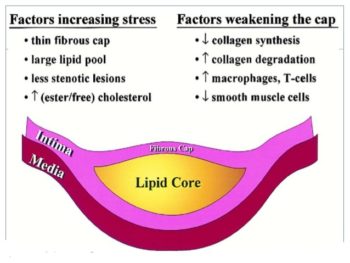
_
Now let us discuss biomechanical factors of plaque disruption:
Heart rate, blood pressure and pulse pressure are all biomechanical factors affecting vulnerable arterial walls, increasing circumferential stress and material fatigue.
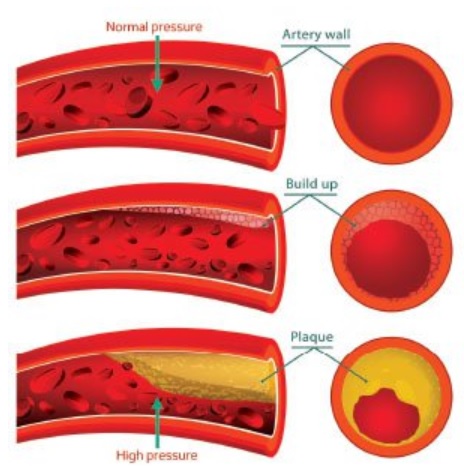
Changes in blood pressure can tilt a plaque from stability into instability and rupture. Many triggers for sudden coronary events have been identified clinically and pathologically: exertion, vigorous exertion, anger and wake-up, and fear. What these all have in common is sympathetic neurohormonal activation responsible for hemodynamic changes, which can rupture vulnerable atherosclerotic plaque. A drop of about 5-6 mmHg diastolic blood pressure is enough to obtain a significant decrease in vascular mortality.
_
Coronary plaques are constantly stressed by a variety of mechanical and haemodynamic forces that may precipitate or ‘trigger’ disruption of vulnerable plaques. Stresses imposed on plaques are usually concentrated at the weak points, namely at points at which the fibrous cap is thinnest and where tearing most frequently occurs.
Tension in Arterial Walls:
The tension in the walls of arteries and veins in the human body is a classic example of Laplace’s law. This geometrical law applied to a tube or pipe says that for a given internal fluid pressure, the wall tension will be proportional to the radius of the vessel.
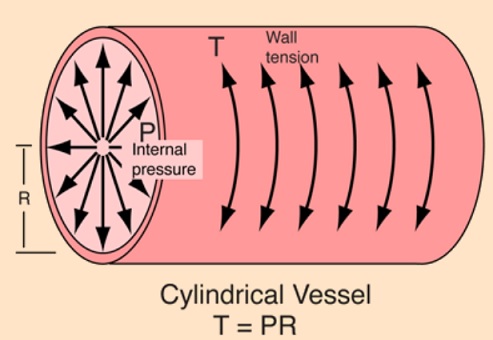
The implication of this law for the large arteries, which have comparable blood pressures, is that the larger arteries must have stronger walls since an artery of twice the radius must be able to withstand twice the wall tension. Arteries are reinforced by fibrous bands to strengthen them against the risks of an aneurysm. The tiny capillaries rely on their small size.
Circumferential wall tension:
Local wall tension major local haemodynamic factor that plays a key role in the pathobiology of plaque rupture. Wall tension is proportional to blood pressure and inversely proportional to lumen stenosis. According to Laplace’s law, wall tension is proportional to the vessel radius for a given blood pressure. Accordingly the tension created in fibrous caps of mildly or moderately stenotic plaques is greater than that created in caps of severely stenotic plaques (smaller lumen) with the same cap thickness and exposed to the same blood pressure. Consequently, mildly or moderately stenotic plaques are generally stressed more than severely stenotic plaques, and could therefore be more prone to rupture. The highest wall stress typically occurs at the upstream shoulders of a stenotic plaque or within a non-stenotic plaque co-localizing with thin fibrous cap, increased macrophage density, and local microcalcifications. Collectively, the dynamic synergism among local haemodynamic factors (ESS and wall stress), plaque composition, and vascular remodeling is a key player in acute plaque disruption. The advent of modern invasive (e.g. 3D OCT) and non-invasive imaging modalities is anticipated to advance our knowledge on these pathophysiological pathways.
Bleeding and/or oedema into plaques from the thin walled new vessels originating from the vasa vasorum and frequently found at the plaque base could theoretically increase the intra-plaque pressure, with resultant cap rupture from the inside. Collapse of severe but compliant stenoses due to negative transmural pressures may produce highly concentrated compressive stresses from buckling of the wall with bending deformation, preferentially involving plaque edges, and theoretically this could contribute to plaque disruption. The propagating pulse wave causes cyclic changes in lumen size and shape, with deformation of plaques, particularly the ‘soft’ ones. Eccentric plaques typically bend at their edges (at the junction between the stiff plaque and the more compliant plaque-free vessel wall). Also, changes in vascular tone cause bending of eccentric plaques at their edges. Cyclic bending may in the long term weaken these points, leading to unprovoked ‘spontaneous’ fatigue disruption, whereas sudden accentuated bending may trigger rupture of a weakened cap.
_
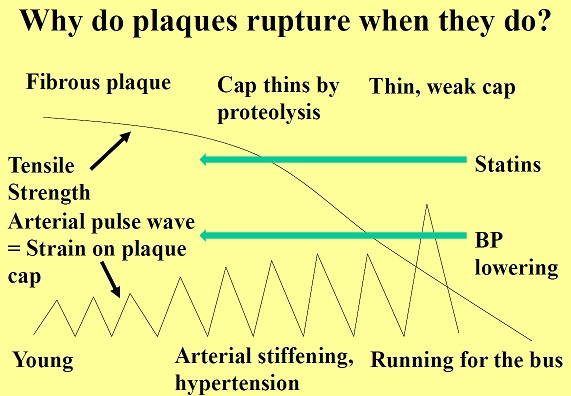
Haemodynamic stresses (ESS) are usually much smaller than mechanical stresses imposed by blood and pulse pressures.
_
The haemodynamic conditions inside blood vessels lead to the development of superficial stresses near the vessel walls, which can be divided into two categories: a) circumferential stress due to pulse pressure variation inside the vessel; b) shear stress due to blood flow. The direction of the shear stress vector is determined by the direction of the blood flow velocity vector very close to the vessel wall. Shear stress is applied by the blood against the vessel wall. On the other hand, the force applied to the blood by the wall is considered as friction and has a direction opposite to the blood flow. The tensions acting against the vessel wall are likely to be determined by blood flow conditions. Shear stresses during turbulent flow, regions of flow recirculation or flow separation, develop more complicated local characteristics. Normal wall stresses due to blood pressure are transferred to all vessel wall layers (intima, media and adventitia). On the other hand, shear stress is applied mainly to the inner layer of the arterial wall in contact with the blood, the vascular endothelium. The normal wall stresses applied (directly), as well as shear stresses (indirectly), regulate blood vessel diameter depending on vascular wall elasticity (distensibility) and endothelial function (endothelial induced vasodilatation), respectively.
_
Shear stress and vascular endothelium:
Endothelium responds to shear stress through various pathophysiological mechanisms depending on the kind and the magnitude of shear stresses. More specifically, the exposure of vascular endothelium to shear forces in the normal value range stimulates endothelial cells to release agents with direct or indirect antithrombotic properties, such as prostacyclin, nitric oxide (NO), calcium, thrombomodulin, etc.
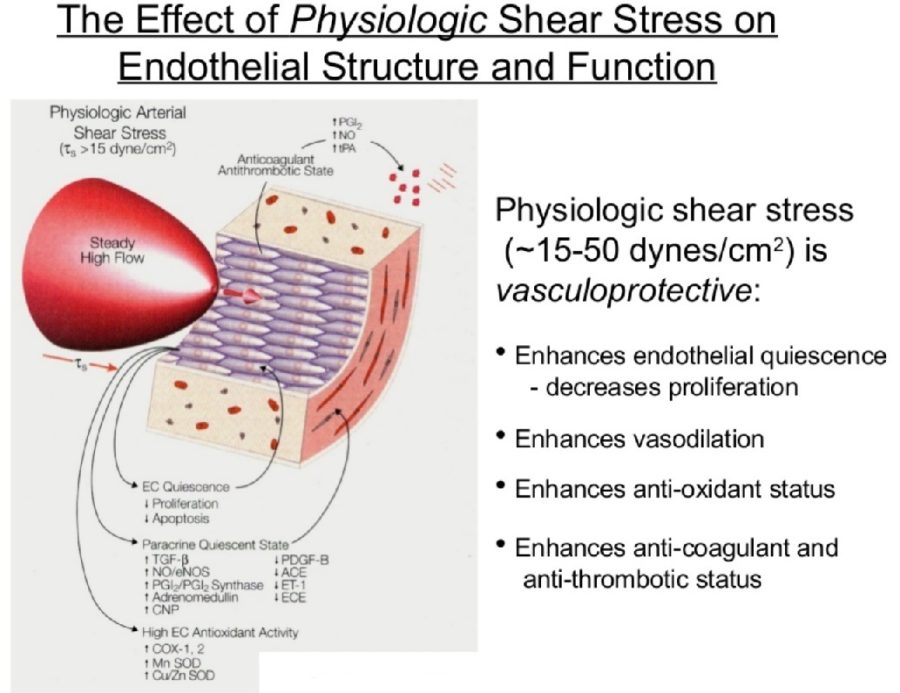
Under normal conditions, shear stresses maintain their direction and their magnitude within a range of values that impedes atherogenesis, thrombosis, adhesion of leukocytes, smooth muscle proliferation and endothelial apoptosis. The arterial and venous vascular tree is exposed to different levels of shear stress, a fact that is determined by flow velocity characteristics. Shear stress at arterial walls ranges between approximately 10 and 70 dynes/cm2, whereas the corresponding normal values for veins are considerably lower, from 1 to 6 dynes/cm2. Higher shear stress values have been observed in the vasculature, especially in regions with an anatomy/ geometry promoting turbulent flow or increased flow velocity (e.g. arteries with strong curvature such as the aortic arch, bifurcations, anastomoses, etc). In those cases, shear stress shows increased values, while the direction may also change due to retrograde flow, which depends on the haemodynamic conditions.
_
Under normal shear conditions, endothelial as well as smooth muscle cells have a rather low rate of proliferation. Changes in shear stress magnitude activate cellular proliferation mechanisms as well as vascular remodelling processes. More specifically, a high grade of shear stress increases wall thickness and expands the vessel’s diameter, so that shear stress values return to their normal values. In contrast, low shear stress induces a reduction in vessel diameter and leads to intima-media hyperplasia. Consequently, shear stresses stimulate vasoregulatory mechanisms which, together with alterations of arterial diameter, attempt to maintain a mean shear stress level of approximately 15 dynes/cm2. The presence of low shear stresses is frequently accompanied by unstable flow conditions (e.g. turbulence flow, regions of blood recirculation, “stagnant” blood areas). Such conditions are often observed in the aorta or at arterial bifurcations. Blood flow patterns are far more variant and complicated at arterial bifurcations including reverse velocity, flow stagnancy, and both high and low shear stress at different locations.
_
Wall shear stress is the tangential drag stress produced by blood moving across the endothelial surface. It is a function of the velocity gradient of blood near the endothelial surface. Its magnitude is directly proportional to blood flow and blood viscosity and inversely proportional to the cube of the radius. Thus, a small change in the radius of a vessel will have a large effect on wall shear stress. Wall shear stress regulates arterial wall remodeling. Arteries enlarge in response to high shear stress. Atherosclerotic plaques localize preferentially in regions of low shear stress as compared to high shear stress according to previous studies. Furthermore, decreased shear stress induces intimal thickening in the vessel which had adapted to high flow. Subnormal shear stress also results in endothelial apoptosis.
_
Endothelial shear stress (ESS) and plaque vulnerability:
Endothelial shear stress, the tangential stress derived by the friction of the flowing blood on the endothelial surface, plays a key role in the pathobiology of atherogenesis, plaque formation, and plaque progression to vulnerability. The implication of ESS in plaque vulnerability has been extensively studied in animal and human studies.
Studies in swine showed that low ESS is a strong stimulus that perpetuates the atherosclerotic process in a dose–response manner leading to plaque vulnerability. Low ESS modulates the accumulation of lipids and neovascularization resulting to plaque volume expansion. In the setting of inflammation, low ESS increases the mRNA expression and activity of major elastolytic MMPs (MMP 2, 9, 12) and cathepsins (cathepsin K and S) relative to their muscle cell apoptosis and reduction in smooth muscle cells and collagen synthesis leads to fibrous cap thinning.
A large, multicentre clinical study investigated the role of ESS in the natural history of atherosclerosis. Five hundred patients with ACS underwent three-vessel IVUS examination and profiling of local ESS at baseline and at 6 months. This landmark study demonstrated that low baseline ESS is an independent predictor of plaque progression and expansive remodelling with lumen narrowing. Data from OCT studies in man further elucidated the role of low ESS in plaque vulnerability. Those studies combined functional and morphological assessment of plaque using 3D OCT and showed that low ESS is associated with inflammation, thin fibrous cap, large lipid core, and expansive remodelling, all of which are key pathobiological fingerprints of vulnerable plaque.
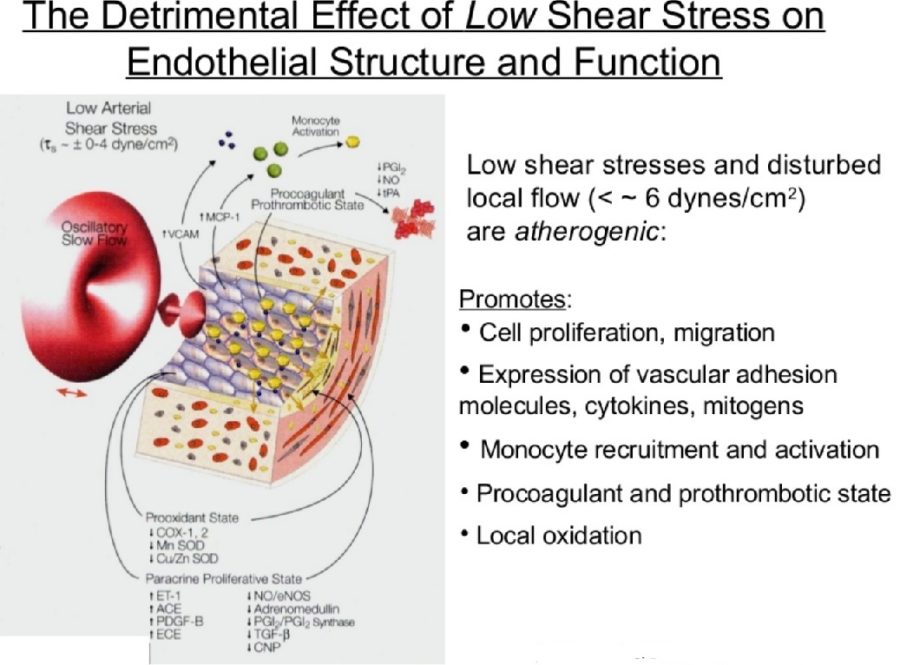
Coronary arteries, carotid bifurcations, abdominal aorta, iliofemoral arteries, the branch points of arteries, and the artery near the curvature are the common sites for atherosclerotic lesion due to the presence of low or oscillatory endothelial shear stress.
_
Endothelial shear stress and plaque rupture:
Local ESS is a major local factor that interplays with the vascular remodeling response, leading to plaque growth and potentially plaque rupture. Non-stenotic vulnerable plaques are exposed to low ESS, which perpetuates an ongoing vicious cycle of local inflammation, matrix degradation, and expansive remodelling leading to acute plaque disruption and thrombotic occlusion of the lumen in the setting of increased thrombogenicity. In contrast to non-stenotic vulnerable plaques, the stenotic vulnerable plaques create a heterogeneous local ESS environment along their length, which involves low ESS in the upstream shoulders, high ESS at the neck of the plaque, and low/oscillatory ESS in the downstream shoulder. The majority of ruptures occur at the upstream shoulders of the plaque, whereas the downstream regions are more stable and therefore less prone to rupture. Plaque rupture can also occur at the most stenotic part of the plaque likely as a result of local erosion in the setting of very high flow and ESS.
_
Figure below shows that when plaques protrude into the lumen, high shear stress (HSS) is formed at the proximal end of the stenosis also whereas low shear stress (LSS) is formed at the distal part.
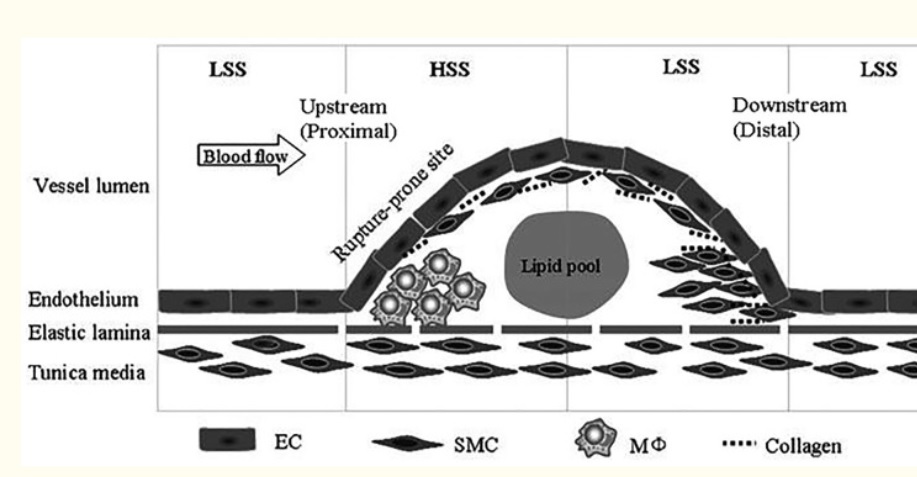
_
High shear stress induces atherosclerotic vulnerable plaque formation through angiogenesis, a 2016 study suggests that although plaque formation is localized in the LSS region, plaque rupture occurs primarily in HSS region. HSS induces up-regulation of NO and VEGF of ECs in the proximal region, which leads to microvessel formation in the plaque from vasa vasorum. Moreover, the pathological angiogenesis is an entry point for infiltration of inflammatory cells, deposition of lipoproteins and the occurrence of intra-plaque haemorrhage. According to this new 2016 study, the magnitude of HSS & LSS and its relationship with atherosclerosis is depicted in table below:
| Term | Location | Magnitude | The relationship with atherosclerosis |
| High shear stress (HSS) | The proximal region of plaque | >25dyn/cm2 | Proathero-sclerotic plaque rupture |
| Low shear stress (LSS) | The distal region of plaque | <10-15dyn/cm2 | Proathero-sclerosis |
In a nutshell:
Both high and low shear stress are atherogenic but plaque rupture is more likely in high shear stress region of plaque according to recent studies. Other studies have shown plaque vulnerability at low shear stress.
_____
_____
Calcification in vulnerable plaque formation and rupture:
Although the degree of coronary calcification is a useful biomarker for the prediction of cardiovascular risk on a population level, the pathogenetic link between the presence of calcification in individual plaques and their potential for rupture is not well established. Conversely, severe calcification in a plaque seems to be a factor associated with stable rather than unstable morphology, While the origin of intimal calcification is not entirely elucidated, it appears that apoptosis of smooth muscle cells and matrix vesicles released by macrophages are key mechanisms in the formation of microcalcifications. Subsequent aggregation of such microcalcifications in larger masses will eventually lead to formation of calcified sheets or plates (macrocalcification), a process that is more pronounced in healed plaque ruptures and fibroatheromas and rarely observed in fibrous plaques. These calcium sheets might later be broken down and form convex calcific protrusions into the lumen with cutting edges called calcified nodules. Calcified nodules, which can be identified by in vivo imaging modalities, are a potential substrate for acute thrombosis.
Several in vivo imaging studies by IVUS or multislice computed tomography have indicated a potential role for calcification in plaque rupture, by demonstrating an association of ‘spotty’ calcification with culprit lesions of ACS. However, pathological studies demonstrated that the amount of calcium in ‘vulnerable plaques’ may vary significantly and that the observed patterns of calcification often resembled other plaque types, thereby limiting the value of spotty calcification as a biomarker of ‘vulnerable plaque’. Recently, a PET-CT imaging study showed better discrimination between culprit and non-culprit plaques in acute coronary syndromes using
18sodium floride (18F-NaF), an imaging radioactive tracer targeting active calcification, rather than 18F-FDG, a non-specific tracer of inflammation. Interestingly, uptake of 18F-NaF was also observed in coronary segments with minimal calcification, while simultaneously several heavily calcified segments had no 18F-NaF uptake. Therefore, 18F-NaF could specifically identify regions with active formation of calcifications, potentially reflecting a process of macrophage-initiated microcalcification deposition. Although microcalcifications, consisting of cellular-size hydroxyapatite depositions within the intima, are located in several locations within the plaque, their presence within the fibrous cap is speculated to be implicated in plaque rupture by compromising the mechanical stability of the plaque. Computational fluid dynamic studies have shown that microcalcifications within the fibrous cap exaggerate the mechanical forces applied to the cap within the cardiac cycle, thus facilitating even thicker cap disruption. Subsequent studies using high-resolution micro-CT have shown that these stresses are exponentially increased in the presence of multiple microcalcifications 0.5 mm in high proximity with each other that can create ‘explosive voids’, allowing for the occurrence of plaque rupture. Nevertheless, the in vivo detection of microcalcifications remains a challenge, as although current imaging modalities, such as OCT, could potentially discern larger microcalcifications, the detection of microcalcifications 0.15–0.20 mm would require higher resolution modalities.
_
Recently, recognized local morphological characteristics of vulnerability (microcalcifications, cholesterol crystals) in combination with local haemodynamic factors (ESS, wall stress) seem to play an important role, while microRNAs are key mediators in plaque destabilization and rupture as seen in the figure below. The identification of new targets for imaging, including microcalcification, cholesterol crystals, and ESS in addition to the local inflammatory environment might enhance the diagnostic accuracy for vulnerable, prone to rupture plaques. These new factors need to be further investigated. 18F-NaF positron emission tomography scan could serve as a concise screening tool. Improvements in computational modelling and simulations can allow for more accurate ESS calculation in a time-efficient manner. Human application of mOCT could help in the identification of macrophage subtypes, microcalcification, and cholesterol crystals. Fusion of modalities, such as OCT with near-infrared autofluorescence, can offer information on both microstructural and molecular characteristics of vulnerable plaques.
_
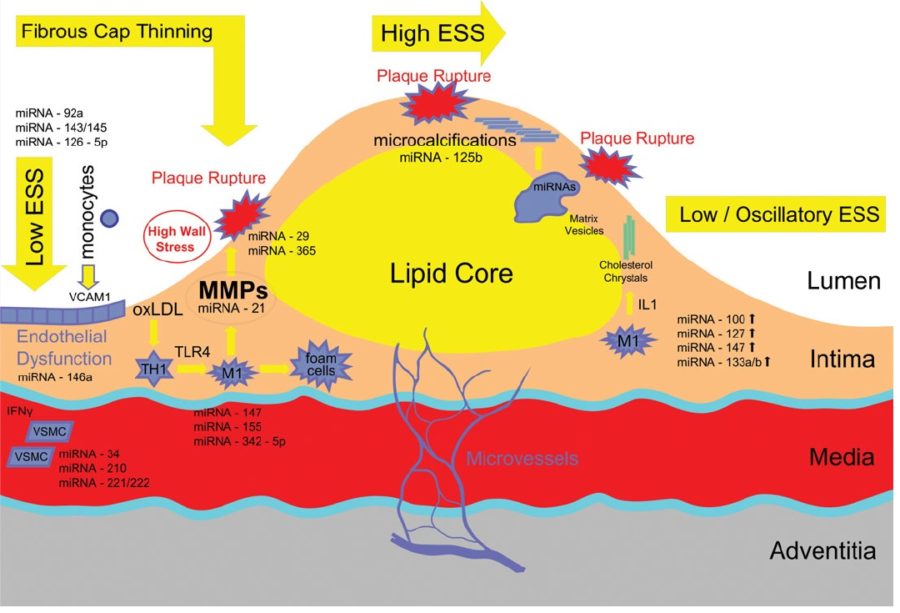
Figure above shows interactions between biomechanical forces, inflammation, microcalcifications, cholesterol crystals, and microRNAs (miRNAs) in high-risk plaque formation and rupture. Atherosclerotic risk factors including dyslipidaemia and low endothelial shear stress at the upstream shoulder of the plaque promote endothelial dysfunction and local inflammation. Recruited monocytes by endothelial adhesion molecules (VCAM-1) are activated within the plaque into M1 macrophages through toll-like receptor 4 (TLR4) and produce matrix metalloproteinases (MMPs), causing the thinning of the fibrous cap. T helper 1 (Th1) cells of innate immunity, activated by oxidized low-density lipoprotein (oxLDL) and other endogenous antigens, regulate the process. Co-localization of high wall stress with cap thinning (upstream shoulders of a stenotic plaque or within a non-stenotic plaque) can lead to plaque rupture. Microcalcifications in high proximity with each other within the plaque can create ‘explosive voids’, allowing for the occurrence of plaque rupture through their interaction with biomechanical forces. Cholesterol crystals through acute volume expansion could also cause mechanical instability or mediate local inflammation through interleukin 1 (IL-1) production. Specific cellular microRNAs regulate these interactions. ESS, endothelial shear stress; ACS, acute coronary syndrome; IFNg, interferon gamma; VSMC, vascular smooth muscle cells.
______
______
Atherosclerotic plaque Angiogenesis:
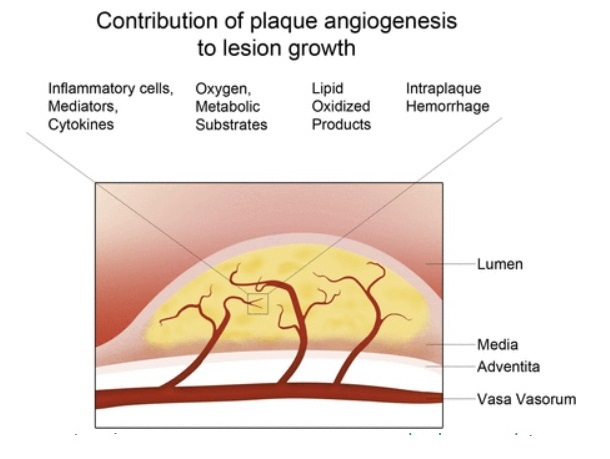
Figure above shows putative role for microvessels in atherosclerotic plaque biology includes augmentation of cellular and soluble lesion components, enhancement of oxygen exchange, delivery of metabolic substrates and lipids, and potentiation of microvascular hemorrhage within the plaque intima.
_
Neovascularization has been recognized as an important process for the progression of atherosclerotic plaques. In fact, vulnerable atherosclerotic plaque prone to rupture are characterized by an enlarged necrotic core containing an increased number of vasa vasorum, apoptotic macrophages, and more frequent intraplaque haemorrhage. Various functional roles have been assigned to intimal microvessels. Angiogenesis increases the supply of oxygen and nutrients to the artery wall and supports initial plaque growth. Once the atherosclerotic plaque develops, intimal angiogenesis is thought to contribute to characteristics of an unstable plaque such as fibrous cap thinning, a cholesterol rich necrotic core, excess infiltration of inflammatory cells, plaque hemorrhage and the increased risk of plaque rupture.
Vasa vasorum angiogenesis which is part of the remodeling process is induced by hypoxia in the early stages of atherosclerosis. Ectopic neovascularization on the other hand occurs in response to an increase in oxygen demand of infiltrated plaque inflammatory cells associated with the late stages of atherosclerosis. Until recently, it was also thought that intimal growth greater than 500 μm reduces oxygen diffusion from the arterial lumen to the vessel wall; however, this has been shown to have little effect on hypoxia. Oxidative stress and local hypoxia stimulate the release of hypoxia inducible factor 1α (HIF-1α) from endothelial cells. In hypoxia, degradation of HIF-1α is inhibited and it enters the nucleus and induces the expression of vascular endothelial growth factor (VEGF), an important factor in the maintenance of endothelial function and integrity. HIF-1α also induces transcription of other pro-angiogenic factors such as platelet derived growth factor (PDGF) and fibroblast growth factor (FGF). The role of VEGF in atherosclerosis is primarily in the formation of new blood vessels. E26 transformation-specific sequence-1 (ETS- 1) is a transcription factor that regulates VEGF-induced angiogenesis. VEGF expression can also be induced in endothelial cells and macrophages by the action of oxidized phospholipids which further stimulate angiogenesis in atherosclerosis.
The role of angiogenesis in atherosclerosis is both adaptive and pathological. Angiogenesis prevents ischemia in early atherosclerosis, however, the morphological characteristics of intra-plaque neovessels and delivery of factors which enhance inflammation and plaque growth suggest angiogenesis contributes to plaque instability. The secretion of matrix melloproteinases during angiogenesis may contribute to fibrous cap thinning as well as the delivery of lipids and reduced the clearance of necrotic cells, increases the growth of the necrotic core. Angiogenesis provides a second route of entry for inflammatory cells into the intima and ultrastructure of plaque neovessels increases the risk of hemorrhage. Angiogenic factors themselves may even contribute to inflammation in atherosclerosis and this places stress and strains on the plaque, increasing the risk of rupture.
______
______
Animal models of atherosclerosis:
The use of animal models of atherosclerosis is an essential tool to improve the understanding of the molecular mechanisms behind atherosclerotic plaque formation and progression, as well as the occurrence of plaque rupture and its associated cardiovascular events. Moreover, animal models allow to assess novel pharmacological treatments that can prevent or slow down the onset of atherosclerosis. In general, animal models for atherosclerosis are based on accelerated plaque formation due to: (1) a cholesterol-rich/Western-type diet, (2) manipulation of genes involved in the cholesterol metabolism, and (3) the introduction of additional risk factors for atherosclerosis, such as diabetes.
An ideal animal model of atherosclerosis resembles human anatomy and pathophysiology and has the potential to be used in medical and pharmaceutical research to obtain results that can be extrapolated to human medicine. Moreover, it must be easy to acquire, can be maintained at a reasonable cost, is easy to handle and shares the topography of the lesions with humans. Mouse and rabbit models have been mostly used, followed by pigs and non-human primates. Each of these models has its advantages and limitations. The mouse has become the predominant species to study experimental atherosclerosis because of its rapid reproduction, ease of genetic manipulation and its ability to monitor atherogenesis in a reasonable time frame. Both Apolipoprotein E deficient (ApoE-/-) and LDL-receptor (LDLr) knockout mice have been frequently used, but also ApoE/LDLr double-knockout, ApoE3-Leiden and PCSK9-AAV mice are valuable tools in atherosclerosis research. However, a great challenge was the development of a model in which intra-plaque microvessels, haemorrhages, spontaneous atherosclerotic plaque ruptures, myocardial infarction and sudden death occur consistently. These features are present in ApoE-/-Fbn1C1039G+/- mice, which can be used as a validated model in pre-clinical studies to evaluate novel plaque-stabilizing drugs.
_
The most commonly used mouse models of atherosclerosis are described in figure below.

Figure above shows overview of current mouse models of atherosclerosis. This figure describes the different models with their total plasma cholesterol levels on normal (ND) and Western-type diet (WD), lipoprotein profile, plaque characteristics, advantages and limitations. The distribution of the plaques in the thoracic aorta and the complexity is shown for mice fed a WD for 20 weeks. The composition of the most complex lesions at that time point is displayed (usually found in the aortic root or brachiocephalic artery).
_
Most important advantages and limitations of commonly used models of atherosclerosis.
| Advantages | Limitations | |
| Mice |
Relatively cheap
Efficient
Easy crossbreeding
Low cost for pharmacological intervention studies |
Differences with human plaques:
Less coronary plaque formation
No intra-plaque neovascularisation and haemorrhage
Rare plaque rupture and thrombosis However, these limitations are overcome in ApoE-/-Fbn1C1039G+/- mice. |
| Rabbits |
Allows translation research such as catheterisation of the aorta (coronary arteries are too small)
Relatively cheap
Easy to breed and handle |
Differences in the localisation of the plaques as compared to humans:
Plaques are most pronounced in the aortic arch and descending thoracic aorta (in contrast to the abdominal aorta in humans) |
| Pigs |
Similarities with human plaques:
Localisation: coronary arteries, abdominal aorta, ileo-femoral
Neovascularisation towards the plaque |
Plaque development mostly ends in the foam cell stage (this can be overcome with the selection of natural mutants, mechanical damage, introduction of other risk factors, genetic engineering with minipigs)
Thrombosis due to plaque rupture is rare (in contrast to humans)
Relatively expensive and more difficult to handle |
| Non-human primates |
Very similar plaque formation as compared to humans (both micro- and macroscopic)
Plaque formation in the coronary arteries |
Non-human primates are expensive and highly regulated
Specialized training is needed |
_
Among the different animal models used to study atherosclerosis, pigs and rats are rarely suitable for exploring plaque neovascularization in the atherosclerotic plaque because they seldom display plaque neovessels (Van der Veken et al., 2016). In contrast, induced advanced atherosclerotic plaques in the thoracic descending aorta of New Zealand white rabbits show intra plaque angiogenesis, as detected using contrast-enhanced ultrasound (Giannarelli et al., 2010).
______
______
Diabetes and vulnerable plaque:
Over the last 30-40 years there has been a gradual decline in mortality from coronary artery disease and this has been attributed to the combination of lifestyle changes and the use of effective cholesterol- and blood pressure-lowering agents, supported by the results of clinical trials. In relation to the specific effects of diabetes, for a number of years there has been a growing opinion that diabetes is a cardiovascular risk equivalent. This view led to the development of the ‘Common Soil Hypothesis’ which stated that diabetes and cardiovascular disease had common antecedents, a notion subsequently supported by studies indicating that this might be the case. More recently, major clinical studies have reported lower than expected rates of ischaemic heart disease on the background of rising rates of diabetes to indicate a dissociation between these two conditions, which sheds doubt on whether diabetes is a cardiovascular risk equivalent. The cause of this finding is not entirely clear, but may indicate the complex benefit endowed by smoking cessation, weight loss and modern therapeutic approaches. Despite, and perhaps because of these observations, the nature of the complex atheromatous lesions associated with the presence of diabetes becomes more important.
_
Pathogenesis of the plaque vulnerability in Diabetes Mellitus:
Atherosclerosis and acute coronary syndrome are more common in diabetes mellitus. Hyperglycemia and hypercholesterolemia in diabetes predispose the arteries to plaque development. Mechanisms such as increased inflammation, foam cell deposition, impaired repair mechanism, endothelial cell dysfunction, vascular smooth muscle cell proliferation, angiogenesis, intra-plaque hemorrhage, and calcification which facilitate the plaque rupture are increased in diabetes mellitus. Thus, diabetes mellitus increases the prevalence of plaque formation and rupture. Diabetes mellitus affects various cellular and molecular effectors involved in plaque development and rupture.
CAD is more common in diabetes mellitus type II (T2DM), or the individuals with persistent hyperglycaemia are more prone to CAD due to increased blood glucose and atheroma formation. Further, it has also been documented that elevated glycosylated haemoglobin and genetically driven hyperglycemia distinctly from T2DM also increases the risk of CAD, suggesting hyperglycemia as a major risk factor for CAD and can affect the pathogenesis as well as the stability of the plaque. Obesity is a chronic inflammatory disease and results in obesity-induced insulin resistance or impaired insulin secretion resulting in hyperglycemia, which further leads to functional and structural alterations of the vessel wall and culminates with diabetic vasculopathies. Hyperlipidemia is another major risk factor for atherosclerosis. Deposition of LDL in the intima initiates the process of atherosclerosis. T2DM is associated with elevated triglycerides, decreased high density lipoprotein (HDL) and increased low density lipoprotein levels collectively characterizing the hyperlipidemia.
Hyperlipidemia and hyperglycemia are the risk factors for atherosclerosis, and increased deposition of lipids and inflammatory cells resulting in necrotic core within the atherosclerotic plaque renders the plaque vulnerable. Nearly 75% of all acute coronary events and 90% of all carotid plaques causing ischemic stroke results due to atherosclerotic plaque rupture. Increased infiltration of inflammatory cells, thin fibrous cap, large necrotic core, and increased angiogenesis are the mechanism involved in plaque rupture. Since hyperglycemia and hyperlipidemia, both associated with T2DM causes the functional and structural alterations of the vessel wall, the core morphological alteration behind atherosclerosis, diabetes mellitus can affect the pathogenesis and process of atherosclerotic plaque formation and rupture.
_
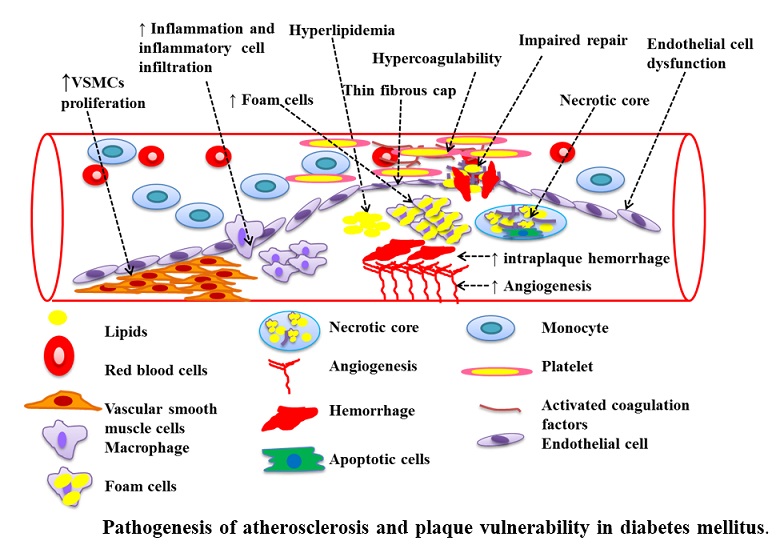
Figure above shows pathogenesis of atherosclerosis and plaque vulnerability in diabetes mellitus. Persistent hyperglycemia in diabetes mellitus potentiate several mechanisms such as hyperlipidemia, increased angiogenesis, intra-plaque hemorrhage, proliferation of vascular smooth muscle cells (VSMCs), infiltration of inflammatory cells, foam cell formation, necrotic core formation due to oxidative stress, and hypercoagulability enhancing the thrombus formation along with thinning of the fibrous cap by altering the collagen content. These mechanisms make the plaque prone to rupture and thereby prevalence of acute coronary syndrome.
_
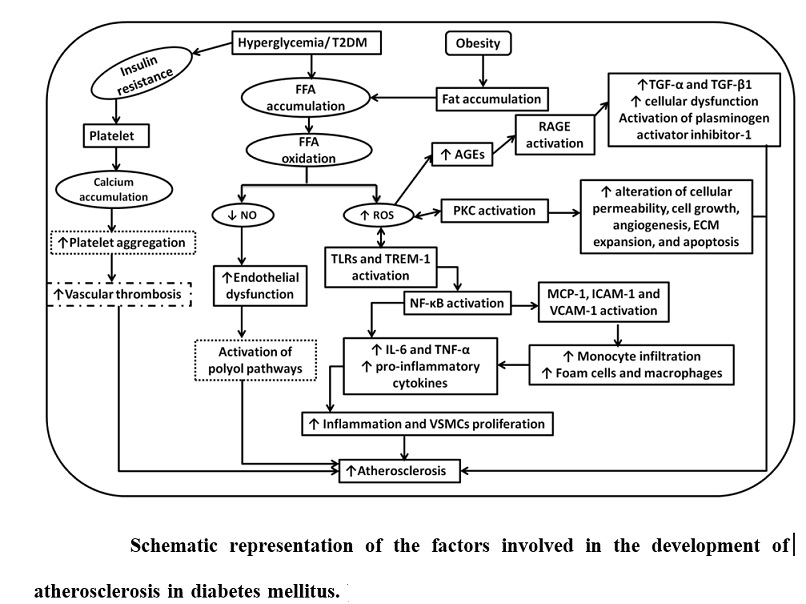
Figure above shows schematic representation of the factors involved in the development of atherosclerosis in diabetes mellitus. Hyperglycemia in diabetes mellitus leads to increased free fatty acid (FFA) accumulation and oxidation. This results in increased oxidative stress due to the generation of reactive oxygen species (ROS) and the decreased availability of nitric oxide (NO). This leads to activation of various inflammatory pathways resulting in increased prevalence of atherosclerosis.
Advanced glycosylation end products (AGEs); diabetes mellitus type 2 (T2DM); extracellular matrix (ECM); interleukin (IL)-6; Intercellular Adhesion Molecule (ICAM) 1; monocytes chemoattractant protein (MCP)-1; nuclear factor kappa beta (NF-kB); protein kinase C(PKC); receptor for advanced glycosylation end products (RAGE); toll-like receptors (TLRs); triggering receptor expressed on myeloid cells (TREM); transforming growth factor (TGF)-alpha (α)-beta (β); tumor necrosis factor (TNF)- α; vascular cell adhesion protein (VCAM)-1; vascular smooth muscle cells (VSMCs).
__
Insulin and atherosclerosis:
Atherosclerotic cardiovascular disease (CVD) is the commonest cause of death in diabetic patients. Several epidemiological studies reported that endogenous hyperinsulinemia is an independent risk factor for CVD, suggesting the possibility of an atherogenic property of insulin. However, hyperinsulinemia is commonly the consequence of compensation for insulin resistance, as in metabolic syndrome, which is itself defined as a cluster of risk factors for CVD. Indeed, other studies showed that insulin resistance is a more potent risk factor for CVD than hyperinsulinemia. Intensive insulin treatment decreased CVD events in patients with type 1 diabetes in the Epidemiology of Diabetes Interventions and Complications (EDIC) study, the follow-up study to the Diabetes Control and Complications Trial (DCCT), whereas the effect of intensive insulin treatment in type 2 diabetes is more controversial. The UK Prospective Diabetes Study (UKPDS) with median duration of follow-up for 10.4 years demonstrated a beneficial effect of intensive insulin treatment to decrease CVD, whereas other trials with shorter follow-up between 3.5 and 6.2 years did not find beneficial effects. Any effect was attributed mainly to changes in glycemic control.
It has been argued whether insulin accelerates or prevents atherosclerosis. Although results from in vitro studies have been conflicting, recent in vivo mice studies demonstrated antiatherogenic effects of insulin. Recent animal studies have demonstrated that impaired insulin signaling by genetic modification accelerates atherosclerosis. In addition, it was shown that insulin treatment reduces atherosclerosis in mice. Taken together, these results indicate protective effects of insulin against atherosclerosis at physiological and pharmacological concentrations, although in most of these studies there were changes in glycemia and other metabolic parameters, which makes it difficult to attribute the effect of insulin treatment to insulin per se. Insulin is a known activator of endothelial nitric oxide synthase (NOS), leading to increased production of NO, which has potent antiatherogenic effects.
______
______
Exercise and vulnerable plaque:
_
The forgotten face of regular physical exercise: a ‘natural’ anti-atherogenic activity, a 2011 study:
Humans are not programmed to be inactive. The combination of both accelerated sedentary lifestyle and constant food availability disturbs ancient metabolic processes leading to excessive storage of energy in tissue, dyslipidaemia and insulin resistance. As a consequence, the prevalence of Type 2 diabetes, obesity and the metabolic syndrome has increased significantly over the last 30 years. A low level of physical activity and decreased daily energy expenditure contribute to the increased risk of cardiovascular morbidity and mortality following atherosclerotic vascular damage. Physical inactivity leads to the accumulation of visceral fat and consequently the activation of the oxidative stress/inflammation cascade, which promotes the development of atherosclerosis. Considering physical activity as a ‘natural’ programmed state, it is assumed that it possesses atheroprotective properties. Exercise prevents plaque development and induces the regression of coronary stenosis. Furthermore, experimental studies have revealed that exercise prevents the conversion of plaques into a vulnerable phenotype, thus preventing the appearance of fatal lesions. Exercise promotes atheroprotection possibly by reducing or preventing oxidative stress and inflammation through at least two distinct pathways. Exercise, through laminar shear stress activation, down-regulates endothelial AT1R (angiotensin II type 1 receptor) expression, leading to decreases in NADPH oxidase activity and superoxide anion production, which in turn decreases ROS (reactive oxygen species) generation, and preserves endothelial NO bioavailability and its protective anti-atherogenic effects. Contracting skeletal muscle now emerges as a new organ that releases anti-inflammatory cytokines, such as IL-6 (interleukin-6). IL-6 inhibits TNF-α (tumour necrosis factor-α) production in adipose tissue and macrophages. The down-regulation of TNF-α induced by skeletal-muscle-derived IL-6 may also participate in mediating the atheroprotective effect of physical activity.
____
Effects of Exercise Training on the Severity and Composition of Atherosclerotic Plaque in apoE-Deficient Mice, a 2011 study:
Aim: To investigate the effects of exercise on atherosclerotic plaque composition, the concentration of matrix metalloproteinases (MMPs) in the atherosclerotic plaque and the systemic circulation.
Methods: Ninety apolipoprotein E-deficient (apoE–/–) mice (45 male) were randomized to the following groups (n = 15 each): control male/female; sedentary male/female; exercise male/female. Mice were kept on a 16-week high-fat diet. Subsequently, the control groups were sacrificed, while the rest of the animals were placed on a normal diet for 6 more weeks. During the latter period, the exercise groups were trained daily on treadmill. At the end of the study, mice were euthanized, and blood samples as well as aortic root specimens were obtained. Results: Compared to control and sedentary animals, exercise training reduced atherosclerotic plaques (–30%; p < 0.01) and increased elastin and collagen content in both genders (p < 0.05). Body weight or lipid profile did not change significantly. Decreased macrophages and MMP-9 as well as increased tissue inhibitor of metalloproteinases 1 (TIMP-1) levels were observed in the atherosclerotic plaques of the exercise-treated groups (p < 0.05). Plasma concentrations of MMP-9 decreased, while plasma TIMP-1 levels increased in the exercise compared to control and sedentary groups (p < 0.05).
Conclusions: Exercise training had a favorable effect on the size and composition of the atherosclerotic plaque in apoE–/– mice, associated with suppressed MMP activity.
In this study, researchers investigated the effects of exercise training on the atherosclerotic plaque size and composition, using a well-documented animal model of atherosclerosis. A significant regression of atherosclerosis after a treadmill training program, independent of body weight reduction or lipid profile modulation, was found. Importantly, these effects were accompanied by favorable alterations in plaque structure, such as an increase in collagen and elastin component, a decrease in macrophage number and a decrease in metalloproteinase activity.
The principal limitation of this study was that advanced atherosclerotic plaques in mice rarely develop into rupture or erosion, the final evolution stage of atherosclerosis in humans. Authors underline that the components of the atherosclerotic plaque (for example, macrophages and collagen) constitute surrogate markers of plaque vulnerability. Despite the observed beneficial changes in those cellular and acellular components, their study did not document direct effects on plaque rupture. Thus, the extrapolation of their results to humans should be done with caution.
____
Plaque Rupture and Sudden Death Related to Exertion in Men with Coronary Artery Disease, a 1999 study:
Regular exertion decreases the risk of coronary heart disease. However, acute exertion may induce significant cardiac events, presumably by precipitating rupture of a coronary artery plaque. This autopsy survey examined the hearts of 141 men with severe coronary artery disease who died suddenly. Severe disease was defined as greater than 75% narrowing of at least one coronary artery. Based on reports of scene investigators, witnesses, or family for witnessed (90) and unwitnessed (51) deaths, the investigators determined that 116 men died at rest, 18 died within one hour of strenuous physical activity, and 7 died within two hours of emotional stress.
Plaque rupture was deemed responsible for 17 of the 25 deaths (68%) during exertion versus 27 of the 116 deaths at rest (23%; P<0.001). Fourteen of the 25 deaths related to exertion occurred in previously sedentary men who engaged in sudden strenuous activity. In men with severe coronary artery disease, sudden death related to exertion was associated with acute plaque rupture.
Vigorous exercise can induce coronary plaque rupture through several triggering mechanisms: increased wall stress due to high blood pressure or increased heart rate and plaque disruption caused by coronary artery spasms or increased flexing of diseased coronary arteries. Additional risk factors induced by high intensity exercise are the higher sympathetic tone and increased thrombogenicity by increased platelet activation contributing to coronary artery thrombosis after a plaque rupture.
On the other hand, a recent study found contrasting results. Causes of exercise-induced cardiac arrest and death were examined in 10.9 million middle-aged and elderly men participating in long distance running races. In patients without cardiomyopathy, autopsy reports suggested oxygen mismatch as primary cause in CAD-induced cardiac arrest, instead of acute plaque rupture. At coronary angiography, not one person with serious CAD (31 runners with complete clinical data, 23 died, 8 survivors) had evidence of plaque rupture or thrombus.
The pathophysiological pathway by which CAD causes cardiac arrest during vigorous exercise is topic of debate. Two pathophysiological mechanisms are possible: (1) Plaque rupture causing myocardial infarction and (2) Demand ischaemia causing ventricular arrhythmias.
_____
Exercise and coronary artery calcium (CAC) studies, 2017:
Exercise is good for you, but is there a point at which more exercise is either no longer beneficial or perhaps even harmful? Two new studies now help clarify — albeit not resolve — the issue. The studies offer evidence that people who exercise for long periods and with great intensity appear to have an elevated risk for atherosclerosis. But, reassuringly, the atherosclerotic lesions that develop in intense exercisers are more likely to be characterized as stable. Earlier studies found that people who exercised a lot were more likely to have indications of atherosclerosis than more moderate exercisers, but the association was clouded because some of the high-volume exercisers were already at elevated risk for cardiovascular disease. The new studies looked at populations of low-risk older athletes.
In the first study, Ahmed Merghani (St. Georges, University of London) and colleagues performed a battery of cardiovascular tests on 152 master athletes and 92 matched controls (70% men). All the participants had a low Framingham 10-year risk score. Although most athletes and controls had normal coronary artery calcium scores (CAC) — 60% and 63%, respectively — male athletes were more likely to have high calcium scores and evidence of coronary plaque. Most (72%) of the plaques in male athletes were calcified, but “their stable nature could mitigate the risk of plaque rupture and acute myocardial infarction,” the authors wrote. By contrast, most (61.5%) of the plaques in controls had a “mixed morphology,” thus indicating that they might be at higher risk for an event. Seven men among the athletes, but none in the controls, had a pattern of late gadolinium enhancement on MRI consistent with a prior MI.
In the second study, Vincent Aengevaeren (Radboud University, the Netherlands) and colleagues studied 284 men with a wide range of lifelong exercise volumes. Overall, 53% of the men had an abnormal calcium score (greater than zero). 68% of men at the highest activity level (greater than 2,000 MET-minutes per week) had an abnormal calcium score, compared with 43% in the lowest activity level group (less than 1,000 MET-minutes per week). As in the first study, plaque type differed among the activity levels. 38% of men in the most active group had calcified plaques compared with 16% in the least active group. “These observations may explain the increased longevity typical of endurance athletes despite the presence of more coronary atherosclerotic plaque in the most active participants,” concluded the authors.
In an accompanying editorial, Aaron Baggish (Massachusetts General Hospital) and Benjamin Levine (University of Texas Southwestern) wrote that “the dose-response relationship between exercise and health outcomes, in particular, at exercise doses that exceed current recommendations, remains incompletely understood.” The two new studies “represent important contributions to the sports cardiology literature but raise as many questions as they answer.” The observational nature of the two studies “is well suited for documenting associations but is not capable of establishing cause-and-effect relationships between exposures and clinical phenotypes,” they wrote, citing the potential role for unadjusted confounding factors. Even more important, they wrote, “it is important to acknowledge the complete absence of clinical outcomes data in athletes with CAC.” Although CAC in less active populations has been linked to adverse outcomes, “it is possible that the presence of CAC among dedicated lifelong endurance athletes may very well represent a clinically benign phenotype.” The editorialists said that physicians evaluating athletic patients should rely on conventional risk factors and do not need to obtain a CAC score except “in limited situations” where it may “provide adjunctive information.”
Asked to comment, Michael Joyner (Mayo Clinic) said that the papers “provide important insight to inform the ‘too much exercise’ debate. The key point is that while some high-volume habitual exercisers have high coronary calcium scores, they also appear to have more stable plaque. Stable plaque plus larger coronary arteries that dilate more in high volume exercisers should be highly protective against coronary events.”
_______
_______
Biomarkers of atherosclerotic plaques:
Plasma markers of vulnerability:
Increased understanding of the processes causing atherosclerosis has facilitated efforts to identify novel markers of risk that may be circulating in plasma and readily available for sampling. Plasma markers of risk that improve the accuracy provided by cholesterol determinations would enhance primary prevention in the general population by identifying individuals in need of further diagnostic testing and would improve secondary prevention by guiding therapy in patients known to have coronary disease. High-sensitivity C-reactive protein (hs-CRP), the most extensively studied plasma marker of inflammation, identifies an increased risk of an acute coronary event independent of LDL cholesterol. Lipoprotein-associated phospholipase A2, another plasma marker associated with inflammation, is an independent predictor of cardiac events. IL-6 is an independent marker of increased mortality in unstable CAD. Soluble adhesion molecules like ICAM-1, VCAM-1, selectins, CD-40 ligand, PAPP-A (pregnancy associated plasma protein A) and markers of immune activation like Anti-LDL antibody, and Anti-HSP (heat shock protein) antibody are among the other biomarkers associated with high risk of an acute plaque event. Nuclear factor-κB, which controls nuclear processes associated with inflammation, is increased in patients with unstable angina. Advances in genetics are already producing markers of vulnerability to coronary events. A markedly beneficial effect of a relatively rare mutation in the gene encoding a protease (proprotein convertase subtilisin/kexin type 9 serine protease) affecting plasma LDL levels was recently reported.
Serological markers of vulnerability:
- Abnormal lipoprotein profile e.g., high LDL, low HDL, and HDL size density, lipoprotein (a) etc.
- Nonspecific markers of inflammation e.g., hs-CRP, CD-40L, ICAM-1, VCAM-1, P-selectin, leukocytosis, and serological markers related to the immune system
- Serum markers of metabolic syndrome e.g., diabetes or hypertriglyceridemia
- Specific markers of immune activity e.g., anti-LDL antibody, anti-HSP antibody
- Markers of lipid peroxidation e.g., ox-LDL and ox-HDL
- Homocysteine
- PAPP-A
- Circulating apoptosis markers e.g., FAS/FAS ligand
_
Serum markers correlated with plaque inflammation:
In recent years, several studies have correlated different serologic biomarkers with cardiovascular disease, leading to a rapid increase in the number of available biomarkers as seen in the figure below:

_
Signature of Circulating MicroRNAs as Potential Biomarkers in Vulnerable Coronary Artery Disease, a 2013 study:
MicroRNAs (miRNAs) play important roles in the pathogenesis of cardiovascular diseases. Circulating miRNAs were recently identified as biomarkers for various physiological and pathological conditions. The circulating miRNA signature, consisting of the miR-106b/25 cluster, miR-17/92a cluster, miR-21/590-5p family, miR-126 and miR-451, may be used as a novel biomarker for vulnerable CAD.
_____
The rationale for the search of biomarkers of vulnerable plaques seems sound. Plaque rupture is a key step in the initiation of an acute coronary syndrome (ACS). The atherosclerotic plaque contains large numbers of inflammatory cells that may release hydrolytic enzymes and cytokines, thereby destabilizing the plaque. Therefore, many inflammatory biomarkers and enzymes have been suggested as potential biomarkers of vulnerable plaques; however, considerable confusion exists in the literature regarding what is meant by a biomarker of plaque vulnerability.
Biomarker of Plaque Rupture:
A biomarker of plaque rupture would show an increasing concentration when a (coronary) atherosclerotic plaque ruptures. A biomarker of plaque rupture would be potentially useful for early diagnosis of acute myocardial infarction (AMI) because plaque rupture precedes myocardial necrosis. Another potential application would be for unstable angina in which plaque rupture, but not myocardial necrosis, occurs. The requirements that a biomarker of plaque rupture must meet to have clinical utility are considerable, however. The biomarker must be abundant in atherosclerotic plaques and be released immediately into the circulation in high concentration after rupture of the plaque. The biomarker concentration must remain increased for quite some time, because plaque rupture might occur up to several days before the patient gets ischemia and thus presents symptoms. Biomarker specificity is another demanding requirement. The biomarker should be not only highly specific for atherosclerotic plaques but also, ideally, specific for coronary plaques only. An important limitation on the use of a biomarker of plaque rupture for early diagnosis of AMI is that a large proportion of all infarctions are not caused by plaque rupture. Autopsy studies have shown that approximately 60% of fatal cases are associated with plaque rupture, and this proportion is probably even lower in real-life situations involving mainly nonfatal AMIs, especially if type 2 AMIs are included. Thus, even with a very good biomarker of plaque rupture, the sensitivity for AMI diagnosis would be limited. On the other hand, plaque rupture might occur without being followed by the formation of a flow-limiting thrombus and the development of AMI. Coronary plaque ruptures have been shown in 7%–27% of stable angina patients and in 3% of patients dying of noncardiac causes. In a study that evaluated 3 biomarkers suggested to indicate plaque rupture [myeloperoxidase (MPO), matrix metalloproteinase 9 (MMP-9), and pregnancy-associated plasma protein A (PAPP-A)] for the early diagnosis of AMI, the diagnostic information was almost nil. For all 3 biomarkers, the areas under the curve in ROC curve analyses were not significantly different from 0.50.
Biomarker of Plaque Instability or Vulnerability:
A biomarker of plaque instability or vulnerability would show increased concentrations in patients who have atherosclerotic lesions in their coronary arteries that are more prone to rupture. Such individuals would thereby constitute a population of individuals at increased risk of cardiac events in the future. A biomarker of plaque instability is intended to be used not for diagnosis but for risk prediction. The concept also implies some sort of mechanistic link between the biomarker and plaque rupture. Otherwise, biomarkers such as cardiac troponin might be considered erroneously as biomarkers of plaque instability, given that troponin increases are associated with an increased risk of a new AMI (i.e., irreversible myocardial cell death). The chain of reasoning is as follows: The biomarker of plaque instability is found in atherosclerotic plaques and is more abundant in plaques with unstable characteristics than in plaques with stable characteristics; an increased concentration of the biomarker in the circulation signals that the patient has plaques with unstable characteristics that are more prone to rupture; and plaque rupture leads to unstable angina or AMI. The results for clinical evaluations of suggested biomarkers of plaque vulnerability are conflicting, however. Generally, these biomarkers seem to be more strongly associated with mortality than with the risk of AMI or major adverse cardiac events, which is illogical given that the biomarkers predict plaque rupture. Furthermore, their associations with prognosis seem to be positive more often when studies test these biomarkers by themselves than when studies evaluate several biomarkers simultaneously. In a study that evaluated multiple biomarkers as indicators of the risk of major cardiac events over a 42-day follow-up for patients admitted with ACS, neither placental growth factor (PlGF) nor lipoprotein-associated phospholipase A2 was found to be predictive as a single biomarker. In another study, MPO, MMP-9, and PlGF were not found to predict the occurrence of a composite of cardiac events within 4 months after an ACS episode, although PlGF taken alone predicted all-cause mortality. In a third study, the biomarkers MPO, MMP-9, and PAPP-A did not predict death or AMI within 1 year, and a fourth study found that none of the biomarkers (MPO, MMP-9, and PAPP-A) predicted the recurrence of a troponin T–positive coronary event or cardiac death within 45 months. Several possible explanations for these negative findings need to be considered, however: A given biomarker might not, in fact, be a biomarker of vulnerable atherosclerotic plaques (e.g., the excretion of PAPP-A from vulnerable plaques has been questioned); inappropriate assays (or sample types) might have been used (e.g., the results for PAPP-A might be different depending on whether free or total PAPP-A was measured); the biomarker might have been measured only at admission when serial measurements might have supplied more information; biomarker results in a stable-angina population might differ from those for a population with ACS; and the biomarker might have been evaluated as part of a single-biomarker strategy rather than as part of a stepwise multibiomarker strategy. Several other questions need clarification before a specific biomarker can be considered useful in a clinical routine. Has the short- and long-term biological variation of the biomarker been characterized? Does the biomarker increase immediately before a plaque rupture, and should we look for a change in concentration over time rather than for the absolute value? Are there differences in predictive value regarding different cardiac events (e.g., between AMI and cardiac death) and in short-term versus long-term follow-up?
The choice of gold standard represents a challenging problem when evaluating a suggested biomarker of plaque vulnerability. Ideally, the gold standard should include a vascular-imaging technique capable of reliably detecting plaque rupture in the coronary artery, such as intracoronary optical coherence tomography; however, such techniques are currently not feasible for large clinical studies of end points.
Biomarker of Vulnerable Patients:
A biomarker of vulnerable patients would be a biomarker demonstrating increased concentrations in patients prone to a new cardiac event (i.e., new AMI). This definition encompasses a much broader concept that does not imply a direct association with the risk of plaque ruptures (e.g., the biomarker could be associated with the risk of flow-limiting thrombus formation once a plaque rupture has occurred). Hence, a biomarker of plaque instability is a biomarker of vulnerable patients, but the opposite is not necessarily the case. In fact most biomarkers that are claimed to be biomarkers of plaque instability or vulnerability indeed belong to this category, and virtually any biomarker that is predictive of new cardiac events could be labeled as a biomarker of vulnerable patients. Thus, the term “biomarker of vulnerable plaque” should be separated from the term “biomarker of vulnerable patient.”
The concept of a biomarker capable of predicting plaque rupture is appealing. A biomarker of vulnerable plaque would allow the possibility to better target preventive measures against plaque rupture and thereby decrease the occurrence of ACS; however, are there really biomarkers of vulnerable plaque? If the question is slightly rephrased to, “Are any useful, scientifically proven biomarkers of vulnerable plaque currently available?” the answer is, unfortunately, “No.” If the question is rephrased to, “Will there be a useful biomarker of vulnerable plaque available in the future?” the answer must be, “Let us hope so, but it is still a long way to get there.”
______
______
Vulnerable plaque imaging techniques:
Rupture of vulnerable plaques may lead to myocardial infarction or stroke and is the leading cause of morbidity and mortality worldwide. Thus, there is a clear need for identifying these vulnerable plaques before the rupture occurs. Atherosclerotic plaques are a challenging imaging target as they are small and move rapidly, especially in the coronary tree. Many of the currently available imaging tools for clinical use still provide minimal information about the biological characteristics of plaques, because they are limited with respect to spatial and temporal resolution. Moreover, many of these imaging tools are invasive. The new generation of imaging modalities such as magnetic resonance imaging, nuclear imaging such as positron emission tomography and single photon emission computed tomography, computed tomography, fluorescence imaging, intravascular ultrasound, and optical coherence tomography offer opportunities to overcome some of these limitations. Among all imaging techniques, intravascular ultrasound (IVUS) and optical coherence tomography (OCT) provide the closest information to match the histopathology of atherosclerotic plaques. However, the invasive nature and high cost of these techniques limit their widespread use. Less invasive techniques, such as MRI or CT, are more appropriate for prevention or screening purposes. Although CT has specific limitations, it provides valuable information that makes it a potentially useful technique for early identification and characterization of coronary plaques. Both CT and MRI can identify certain unstable plaque characteristics thought to be associated with an increased risk of rupture and events. PET imaging allows the activity of distinct pathological processes associated with atherosclerosis to be measured, differentiating patients with inactive and active disease states. Hybrid integration of PET with CT or MRI now allows for an accurate assessment of not only plaque burden and morphology but plaque biology too.
_
Emerging Techniques for Detection of Vulnerable Plaque:
- Invasive Techniques:
Angioscopy
Intravascular Ultrasound (IVUS)
Intravascular Thermography
Intravascular Optical Coherence Tomography (OCT)
Intravascular Elastography
Intravascular and Transesophageal MRI
Intravascular Nuclear Imaging
Intravascular Electrical Impedance Imaging
Intravascular Tissue Doppler
Intravascular Shear Stress Imaging
Intravascular (Photonic) Spectroscopy
-Raman Spectroscopy
-Near-Infrared Diffuse Reflectance Spectroscopy
-Fluorescence Emission Spectroscopy
-Spectroscopy with contrast media
Intra-coronary assessment of endothelial function
Intra-coronary measurement of MMPs and cytokines
- Non-Invasive Techniques:
MRI
-MRI without contrast media
-MRI with contrast media: Gadolinium-DPTA
-MR Imaging of Inflammation: Super Paramagnetic Iron Oxide (SPIO and USPIO)
-MR Imaging of Thrombosis using monoclonal Ab
Electron Beam Computerized Tomography (EBCT)
Multi-Slice Fast Spiral / Helical Computed Tomography
Nuclear Imaging (18-FDG, MCP-1, Annexin V, CD40)
- Blood Tests / Serum Markers:
CRP, ICAM-1, VCAM, p-Selectin – Proinflammatory cytokines – Lp-PLA2 – Ox-LDL Ab – PAPP-A
- Endothelial Function Test:
-Intra coronary acetylcholine test
-Noninvasive flow mediated dilatation of brachial artery
-Anti-body against endothelial cells
_
Although sampling of peripheral blood to assess vulnerability has the advantages of ease of use and low cost, imaging the actual extent of atherosclerosis in vessels provides improved risk stratification. For primary prevention, emphasis is on non-invasive imaging of vessels in those deemed to be at higher risk after initial assessment with demographic characteristics and plasma markers. For secondary prevention in patients already undergoing catheterization, invasive imaging can be performed with minimal added risk and cost and has considerable advantages over non-invasive imaging.
_
Figure below shows advances in vulnerable plaque detection chronologically:
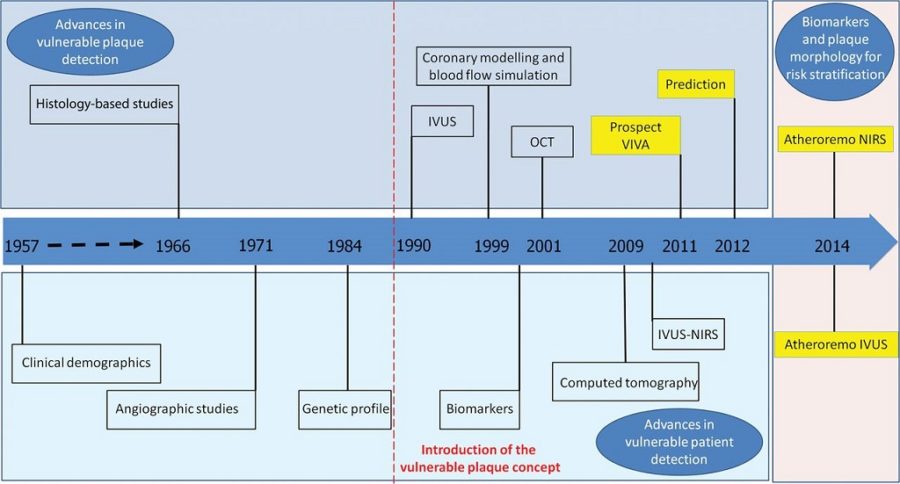
_
Although the treatment of acute coronary syndromes (ACS) has advanced considerably, the ability to detect, predict, and prevent complications of atherosclerotic plaques, considered the main cause of ACS, remains elusive. Several imaging tools have therefore been developed to characterize morphological determinants of plaque vulnerability, defined as the propensity or probability of plaques to complicate with coronary thrombosis, able to predict patients at risk. By utilizing both intravascular and noninvasive imaging tools, indeed prospective longitudinal studies have recently provided considerable knowledge, increasing our understanding of determinants of plaque formation, progression, and stabilization. The current understanding of modes of progression of atherosclerotic vascular disease and the appropriate use of available diagnostic tools may already now gauge the selection of patients to be enrolled in primary and secondary prevention studies. Appropriate trials should now, however, evaluate the cost-effectiveness of an aggressive search of vulnerable plaques, favoring implementation of such diagnostic tools in daily practice.
______
Non-invasive Imaging Modalities:
Non-invasive imaging techniques do not only visualize the plaque but also could gather data on intraplaque haemorrhage (IPH), plaque inflammation, calcification, and plaque remodelling, thus providing the examiner with information regarding the degree of plaque vulnerability. The non-invasive modalities include computed tomography (CT), magnetic resonance imaging (MRI), ultrasound (US), positron emission tomography (PET), single positron emission computed tomography (SPECT), and microwave radiometry.
Table below shows Non-invasive imaging modalities to detect a vulnerable plaque:
| Non-invasive imaging technique | Spatial resolution | Plaque characteristic identified | Advantages | Limitations | |
| CT | 50 micron | Plaque morphology (eccentric pattern, outward remodelling, and spotty calcifications), coronary plaque burden, cap thickness, and macrophages (N1177-specific contrast agent) | High spatial and temporal resolution, real time, quite fast, operator-independent, and excellent calcium detection | Radiation exposure, contrast, difficult to distinguish thrombus, blooming artefacts by calcium, and claustrophobia | |
| MRI | 10–100 micron | Plaque morphology, plaque composition, lipid-rich necrotic core, intraplaque haemorrhage, neoangiogenesis, macrophages, flow measurement, and quantification of stenosis | No radiation, high soft tissue contrast, can be repeated over time, functional, operator-independent, with or without contrast, and many plaque components detected | Low resolution, system fibrosis due to contrast agent, time-consuming, metal implants contraindicated, claustrophobia, cardiac motion artefact, and limited spatial resolution | |
| Ultrasound | 50 micron | Plaque morphology, intima media thickness, flow velocities, and neoangiogenesis (contrast US) | High temporal resolution, cheap, easy to use, no radiation, bedside/large availability, fastest, and functional | Limited sensitivity and specificity, interobserver variability, calcium and air artefacts, limited spatial resolution, and penetration | |
| PET | 1-2 millimeters | Plaque inflammation, macrophages, and neoangiogenesis | High sensitivity and specific targets are detected | Limited resolution, radiation exposure, expensive, limited availability, myocardial uptake of FDG, and cardiac motion artefact | |
| SPECT | 0.3–1 millimeters | Plaque inflammation, apoptosis, lipoprotein accumulation, chemotaxis, angiogenesis, proteolysis, and thrombogenicity. | High sensitivity, low cost, and more spatial resolution as compared with PET | Limited resolution, nonspecificity, radiation exposure, limited availability, and cardiac motion artefact |
_____
_____
Invasive (intravascular) Imaging Modalities:
Invasive imaging techniques utilize intravascular catheters that are mounted with an imaging device and have revolutionized our understanding of the atherosclerotic plaque. Invasive imaging modalities are able to provide the highest resolution images with in-depth analysis about vessel wall and plaque morphology. The current intravascular imaging techniques available to assess vulnerable plaques include intravascular ultrasound (IVUS), optical coherence tomography (OCT), intravascular magnetic resonance, and near infrared spectroscopy (NIRS).
Table below shows Invasive imaging modalities to detect a vulnerable plaque:
| Invasive imaging techniques | Spatial resolution | Plaque characteristic identified | Advantages | Limitations |
| IVUS | 150–250 micron | Plaque distribution, severity, cross-sectional area, and characterization of plaque (lipid core and spotty calcification) | High resolution images of vessel wall and plaque structure | Intra- and interobserver subjectivity, invasiveness, limited spatial resolution, and limited temporal resolution |
| OCT | 4–20 micron | Plaque composition (fibrous, fibrofatty, and fatty), thin fibrous cap, macrophages, neoangiogenesis, and collagen formation | 10 times higher image resolution compared to IVUS and greater tissue contrast | Requires blood-free imaging field, intra- and interobserver variation, invasiveness, and limited tissue penetration |
| IVMR | 120 micron | Early atherosclerosis and more advanced plaque formations and plaque composition (lipid, fibrous, and calcified tissues) | High resolution of plaque structure and composition | Invasiveness and need for occlusion balloon |
| NIRS | NA | Thin fibrous cap, lipid core, and macrophages | High resolution of plaque structure with reliability | Invasiveness, limited tissue penetration, and cardiac motion artefact |
______
______
Overview of vulnerable plaque detection by imaging:
Despite significant advances in imaging, diagnosis of vulnerable plaque remains problematic. Most current techniques identify luminal diameter or stenosis, wall thickness, and plaque volume but remain unable to identify high-risk plaques.
_
Tools for imaging the vulnerable plaque:
_
Figure below shows significant stenosis in the left anterior descending artery visualized by different imaging techniques: (a). Coronary angiogram revealing (arrow). (b). Intravascular ultrasound with three-dimensional reconstruction showing severe calcification (upper quadrant of transverse image; brackets in longitudinal image). (c). Three dimensional cardiac imaging using multislice computed tomography and (d). Maximum intensity projection of three-dimensional cardiac multislice computed tomography showing severe calcification (arrow).
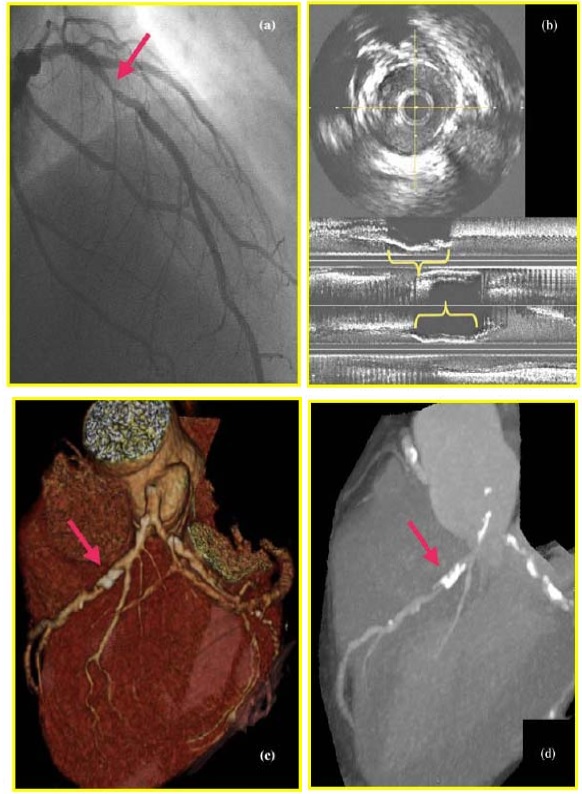
_
Although the ability of imaging modalities to visualize atherosclerotic plaques has improved considerably during the past few years, they primarily focus, with few exceptions, on accurately depicting a still frame of the plaque structure, not the continuous and variable interaction with its surroundings (plaque‐centered approach). Thus, none of them has, beyond the context of proof‐of‐concept studies, established criteria for assessing plaque vulnerability in the broader sense.
Intravascular ultrasound (IVUS) is 1 of the 2 most widely used invasive coronary atherosclerotic plaque imaging modalities, the other being optical coherence tomography. Essentially a catheter‐mounted ultrasound transducer, it follows the same principles regarding ultrasound generation, signal reception, data processing, and image presentation. A grayscale image is generated based on which plaques can be broadly classified as (their echogenicity compared to that of the fibrous adventitia) soft, intermediate, calcified, and mixed. Moreover, added modules can enhance tissue characterization abilities of IVUS, allowing for detection and quantification of different plaque structures—this is achieved, in broad, by analyzing, in addition to reflected signal amplitude, its frequency and power. Virtual histology IVUS, iMAP IVUS, and integrated backscatter IVUS are examples of this, with high reproducibility although they remain proprietary and use different color‐coding for the same structures.
Regarding vulnerable plaques, IVUS, and its modular expansions, can reliably assess plaque burden, expansive remodeling, as well as presence and relative proportion of necrotic core, calcifications, and neoangiogenesis (contrast enhanced; penetration depth, 5 mm). Confirmation of greater strains in fibroatheromatous, as compared to fibrous plaques, has also been provided by compound ultrasound strain imaging (alternatively known as palpography/elastography). However, at its current form (20/40 MHz transducers), it cannot visualize plaque caps, especially thinner ones (resolution, >100 μm). Most prospective studies regarding ability of vulnerable plaques to predict events used IVUS‐based approaches in their definition of vulnerability.
Optical coherence tomography (OCT) is the second major coronary plaque invasive imaging modality that uses near‐infrared light (1.3 μm in wavelength) in a manner analogous to sonography. Light reflected form plaque structures provide image data whereas the background effect of scattered light is negated through use of interferometric techniques. Bright or dark areas occur as a result of constructive or destructive interference between reflected and reference beams. Given the much smaller wavelength of light in comparison to ultrasound, OCT resolution is 1 order of magnitude higher (≈10 μm), allowing for proper visualization of fibrous caps, collagen content (polarization‐sensitive OCT), macrophages (related to inflammation), neovessels (appearing as microchannels), ruptures, and thrombi. OCT performance, with regard to microcalcifications at the lower end of the spectrum (<5 μm in diameter), remains uncertain. Consequently, several, but not all, aspects of vulnerability are visualized. However, this comes at a price, considering that high scattering of light prevents imaging of plaque structures at deeper layers (penetration depth, 1.5 mm) and thus proper recognition and estimation of remodeling and necrotic core size. OCT has displayed high agreement with histopathology regarding both plaque characterization and cap thickness.
OCT limitations include the need for blood displacement, attributed to the high scattering effect of its components, longer image acquisition times (although newer, frequency domain analysis‐based processing methods), frequent artifacts, inability to assess deeper plaque structures, as well as relatively poor discrimination between calcified areas and lipid core, given that both appear as signal poor areas (with clear and diffuse borders, respectively).
A newer form of OCT, micro OCT (μOCT), is currently able to offer axial and lateral resolution in the order of 1 to 2 μm, by means of advanced frequency domain analysis and use of broad bandwidth light, approaching histology levels. Thus, events in the cellular and molecular level, crucial to atherosclerosis development and progression, are visualized, including leukocyte diapedesis, fibrin strand formation, ECM production, endothelial denudation, microcalcifications and cholesterol crystal formation, and penetration of the fibrous cap. Given that this method may, in the future, offer real‐time in vivo images (as well as 3‐dimensional reconstruction) of the vessel wall at subcellular resolution, it may prove extremely useful for the classification of atherosclerotic plaques and identification of the vulnerable ones, replacing classical histology and scanning electron microscopy, notwithstanding all inherent classical OCT limitations.
_
Many arrows in the quiver:
Cardiologists have so many imaging modalities, and every one of them has plus and negative points as seen in the figure below:
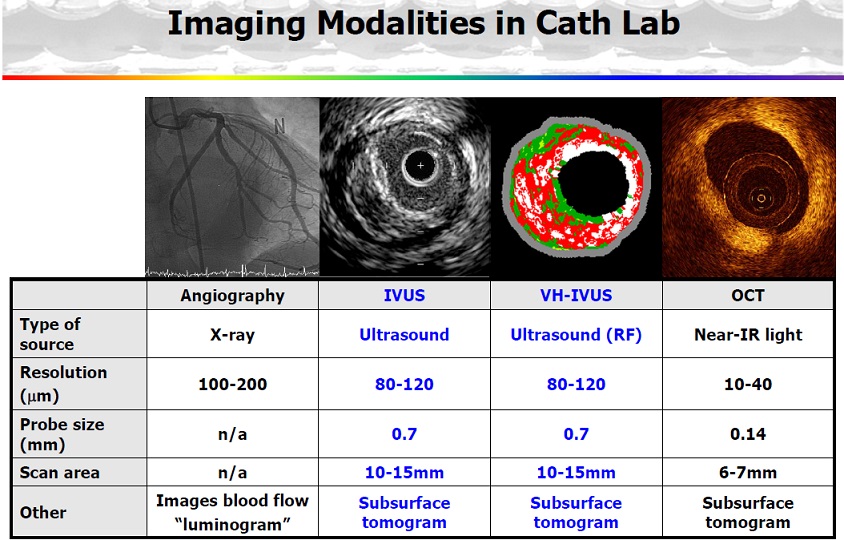
_
Invasive coronary thermography is an approach aiming to detect subtle temperature increases of the vessel wall in areas of heat production, usually accompanying inflamed and/or ruptured atherosclerotic plaques. Purpose‐built catheters include hydrofoil and balloon‐based designs, ensuring adequate apposition of the thermistor module on the vessel wall. The latter is furthermore able to ensure temporary lumen occlusion, thus negating any cooling, convection‐based effects of blood flow, and accentuating underlying gradients. Indeed, studies have shown both an increase in temperature correlating with presenting disease severity (stable angina<unstable angina<acute MI), and a correlation with systemic levels of inflammatory mediators, such as C‐reactive protein. In addition, prognostic value of postangioplasty measured plaque temperature has been demonstrated, given that a difference of ≥0.5°C between plaque and healthy vessel wall was associated with 41% probability of adverse events (repeat infarction, recurrent angina, and death) during a follow‐up period of just 18 months. Good correlation with IVUS‐derived indices of vulnerability (positive remodeling) has been reported. Despite a sound pathophysiological basis, technical issues have, so far, precluded the use of thermography for plaque vulnerability assessment.
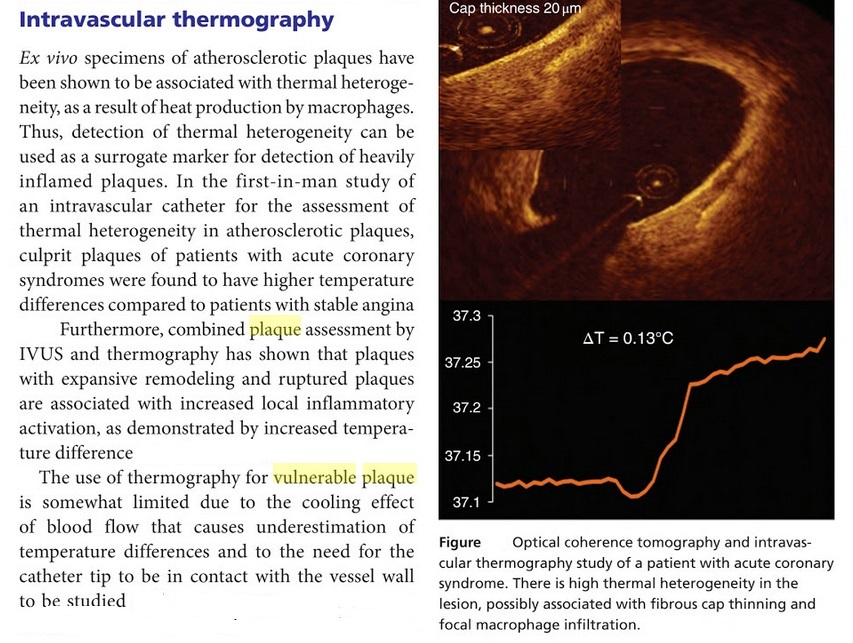
_
Microwave radiometry (MR) is a newly developed, noninvasive method, which possesses a high level of accuracy in the detection of the relative changes in temperature of human tissue, thus indicating degree of inflammation within an atherosclerotic plaque. Both experimental and clinical studies have proved the efficacy of microwave radiometry in the detection of vulnerable plaques, with recent studies also demonstrating the association of microwave radiometry with plaque neovascularization as assessed by contrast enhanced ultrasound (CEUS). After validation of MR in rabbit studies as test subjects, the first application of MR in human carotids was performed to demonstrate its utility in the detection of thermal heterogeneity. Forty-four patients with significant carotid artery stenosis were included in the study. The primary outcomes of this study showed that MR can measure thermal heterogeneity of carotid atheromatic plaques in vivo and that in vivo temperature measurements by MR correlated well with the ultrasound findings of atherosclerotic plaque characteristics. There is no gold standard method for the in vivo quantification of neovascularization and/or inflammation detected by MR; thus more clinical studies are needed.
_
Near‐infrared spectroscopy (NIRS) utilizes characteristic emission spectra produced by plaque contents following interaction with photons (wavelength area, 700–2500 nm). Low sensitivity, in terms of induced response, and high penetration, as compared with visible light spectroscopy, render this method appropriate for assessing the lipid content of plaques, especially in cases of positive remodeling, with large, deep‐seated, lipid‐laden necrotic cores. Studies using NIRS have shown that large lipid content, rather than plaque burden, is associated with thin cap fibroatheroma features. Larger lipid core burden has been shown to accurately differentiate between culprit and nonculprit lesion in ST‐elevation MI patients and has been associated with higher risk for periprocedural myocardial infarction. Interestingly, combination with IVUS may allow for concomitant appreciation of both plaque structure and composition, comparing favorably with OCT. Despite recent studies linking lipid‐rich, NIRS‐defined non-culprit plaque presence with a 4‐fold risk for adverse events (all‐cause mortality, nonfatal ACS, stroke, and unplanned revascularization—excluding those definitely related to the initial culprit lesion) within the first year of follow‐up, it cannot be inferred whether these were actually triggered by the detected vulnerable plaque (thus being amenable to preventive stenting) or by other, not assessed lesions (NIRS was only performed over a vessel segment). The latter could have very well been nonvulnerable lesions at the examination time, and their progression would reflect the need to reduce the atherosclerotic burden as a whole, rather than perform localized treatment. Results from the COLOR and LRP (Lipid Rich Plaque) studies are expected to shed light on the role of NIRS in guiding non-culprit vulnerable plaque stenting.
A modified form of NIRS, near‐infrared autofluorescence, involves active stimulation of lipid components to emit detectable infrared light. Combination with OCT has allowed for better visualization of lipid‐laden necrotic cores and, moreover, accurate localization of the area within it with the densest macrophage concentration.
Computed tomography coronary angiography (CTCA) [Coronary computed tomography angiography (CCTA)] utilizes a computed tomography (CT) scanner in combination with iodine intravenous dye to visualize the coronary arteries. Significant advantages stem from its very nature: noninvasiveness, ability for imaging the whole coronary vasculature, and potential for assessing both vessel wall in addition to the lumen. Advances in technology have allowed for improved image quality with reduced scan times and radiation exposure. More specifically, multislice (or multidetector) scanners possess a 2‐dimensional detector array allowing for simultaneous acquisition of multiple planar images (slices), reducing examination time and possibility for motion artifacts—those with an array breadth of 160 mm allow for imaging of the heart in a single beat. Use of softer reconstruction kernels leads to improved soft‐tissue visualization, however at the expense of spatial resolution. Prospective gating can thus be used in order to synchronize image acquisition with diastole (increased coronary blood flow). Moreover, advent of dual‐energy monochromatic scanners may overcome limitations in tissue assessment caused by adjacent intense calcification, by negating the beam‐hardening effect. Finally, dual‐source scanners may complete the examination in only half a rotation. The related, albeit simpler, technique of calcium scoring does not use an intravenous dye and focuses primarily on coronary calcium quantification to predict the risk for subsequent events.
From a clinical standpoint, CTCA has emerged as the best noninvasive imaging modality in terms of assessing both lumen area and plaque composition, throughout most of the coronary vasculature (vessels >1.5 mm in diameter, where the majority of plaques are located). Its accuracy in detecting lesion presence is noninferior to IVUS, and, consequently, it exhibits an excellent rule‐out ability with a negative prognostic value approaching 100%, even in prolonged follow‐up. Furthermore, although unable to quantify cap thickness, it is nevertheless able to detect presence of thin cap fibroatheromas, comparing favorably with OCT.
Regarding vulnerable plaques, CTCA can reliably assess the presence, size, and thickness of the necrotic core, by grading tissue in Hounsfield units (HU; plaques with large cores will cause less attenuation and thus have lower unit values). Specific high‐risk plaque criteria have been developed, such as positive remodeling (remodeling index [RI], ≥1.1, also a surrogate for plaque composition, (very) low (given that the threshold for a ≥10% necrotic core is 41 HU) attenuation plaque (LAP; <30 HU), napkin‐ring sign (a low attenuation core surrounded by a rim of relatively high attenuation—pathogenesis is still debatable), and presence of spotty calcifications (<3 mm—in accord with the role of calcium specs in destabilizing plaques).
It is noteworthy that all of its derived parameters regarding plaque characterization have emerged as significant and independent predictors of events in multiple studies. For example, a recent coronary CT study, with a follow‐up of almost 100 months, confirmed that, in nonobstructive disease, positive remodeling, increased plaque burden, low attenuation, and napkin‐ring sing presence are all associated with cardiac events. In addition, a plaque volume of 3 mm-cube/mm of vessel wall has been identified as an appropriate cutoff for prediction of adverse events. Presence of multiple high‐risk features has been found to confer a more‐than‐additive risk, as reported in large studies. More specifically, presence of both positive RI and LAP yielded a more than 22% probability of ACS over an average follow‐up of 27 months, as compared to <0.5% probability, should both features be absent. Presence of 3 high‐risk features (LAP, RI, and napkin‐ring sign) leads to a 60% survival free from ACS in the first year. In the latter study, the napkin‐ring sign had the highest hazard ratio (HR) for occurrence of events (HR, 5.6).
Coronary artery calcium score (CACS) is an alternative parameter measured by means of cardiac CT without the need for intravenous dye. Despite the fact that it only assesses 1 aspect of atherosclerosis, as opposed to CTCA, it has been proposed that the latter offers additional prognostic information only in patients with intermediate to high CACS. In another study, a combination of traditional risk factors, CACS, and CTCA yielded an area under the curve of 0.93 for the prediction of major adverse cardiovascular events (cardiac death, nonfatal infarction, and revascularization) over a follow‐up of ≈1000 days, significantly higher than that of the CACS—risk factors combination (0.82; P<0.001).
The noninvasive nature of CTCA renders it an attractive option for population screening (primary prevention), and the ability to assess and monitor plaque burden essentially in the totality of the coronary arterial bed, with incremental improvement over conventional stratification, allows for the pursuit of risk‐factor–reducing strategies as an alternative to pre‐emptive stenting of high‐risk asymptomatic lesions. CTCA‐based 3‐dimensional reconstruction of vessel anatomy and geometry has also been used for the noninvasive assessment of fractional flow reserve in order to determine hemodynamic significance of lesions; however a potentially more‐fruitful approach could be its integration into fully fledged computational models for the assessment of plaque stability.
Magnetic resonance imaging (MRI) has the best ability to visualize the soft‐tissue component of atherosclerotic plaques, as well as neovessel formation and diffusion properties (wall permeability). Major limitations include motion artifacts and use in cases of cardiac implants or devices. Importantly, MRI has been used in animal studies attempting to prospectively determine vulnerable plaque features specifically related with (pharmacologically induced) disruption. These were related to plaque remodeling (with positive remodeling and grater plaque area associated with future rupture) and inflammation indices (markedly increased gadolinium enhancement—denoting increased neovessel permeability and extracellular space expansion—as in intense apoptosis/necrosis). Even so, applicability to humans, the time frame between feature presence and (non-triggered) rupture and relation with clinical events remains unknown.
Molecular imaging involving targeting and visualizing specific components of biological processes, has been extensively used in attempts to visualize the vulnerable plaque. Theoretically, any modality may be used for molecular imaging as long as a proper tracer can be developed, for example, photon‐emitting or possessing paramagnetic properties. Any substance of interest may act as a target provided that it can be either attached to or modified as a tracer.
Positron emission tomography (PET) and single‐photon emission computed tomography (SPECT) are the 2 modalities mostly associated with molecular imaging, often in conjunction with CT/MRI imaging, to provide anatomic background and resolution on top of the mainly functional results of molecular imaging.
Virtually every atherosclerosis‐related process has been studied by means of molecular imaging. More important, vulnerability‐related processes have proven amenable to imaging, including leukocyte adhesion (through involved proteins, such as selectins and vascular cellular adhesion molecule 1), macrophage content (osteopontin), collagen degradation (labeled matrix metalloproteinase [MMP] inhibitors), cell apoptosis (use of annexin that binds to lipids exclusively present in the outer layer of apoptotic cell membrane) or necroptosis (radiolabeled necrostatin, a preferential inhibitor of necroptosis), and neoangiogenesis (use of labeled anti–vascular endothelial growth factor antibodies as tracers). PET has also been experimentally evaluated for the study of intraplaque hypoxia and assessment of macrophage content. Significance of tracer choice can be perceived in the case of plaque calcification, where PET‐CT use of radioactive sodium fluoride as a tracer may reveal sites of active calcification, thus differentiating between stabilizing (encasing) and destabilizing (ongoing) calcification. This is achieved because of preferential binding of the tracer on hydroxyapatite crystals, whose exposed surface is much larger in areas of ongoing crystal formation. PET is generally favored over SPECT because of lower artifact rates and improved spatial resolution as a result of detection of 2, as opposed to a single, oppositely moving photons.
The advent of nanotechnology has further widened the repertoire of imaging probes, allowing the construction of complex nanostructures (nanoparticles and magnetic nanoparticles), engineered according to the desired targets. For instance, nanoparticles composed of cross‐linked iron oxide molecules and 19F perfluorocarbon are used to detect macrophages and in MRI spectroscopy, respectively (the latter attributed to excellent background noise suppression—no fluoride isotopes exist in tissues). Many of these probes may double as treatment vectors (theranostics), inasmuch as under the influence of infrared lasers from a catheter they overheat and release energy to the surrounding tissue, leading to (localized and targeted) irreparable damage (nanobombs). Interestingly, different approaches to optical microscopy (Brillouin microscopy) could allow for direct estimation of plaque stiffness without need for computational models (see below). More specifically, they exploit differential energy shift of emitted photons following interaction with different areas of an anisotropically deformed (strained) structure (such as a plaque cap). Proof‐of‐applicability studies have been undertaken in an ex vivo setting, providing encouraging results regarding potential mounting of Brillouin scattering systems on coronary catheters.
Computational models will allow for simulation of plaque behavior under (patho)physiological conditions. These include finite element analysis models, assessing forces applied on a surface though numerical approximation of the solution by dividing the surface into multiple (>10,000) elementary components with homogeneous properties, and fluid‐structure interaction simulations, also integrating rheological effects on plaque structural stress. They are the only means able to actually assess true vulnerability by integrating the plaque structure with its surroundings; however, all of them are critically dependent on an accurate imaging technique to provide vessel geometry and plaque structural data as input. Intrinsic parameters classically associated with vulnerability (cap thickness, necrotic core size, vessel anisotropy, and all forms of calcification) are combined with the anticipated effects of applied stresses, fatigue attributed to vessel motion during the cardiac cycle, and blood pressure/flow effects to yield meaningful results regarding conditions leading to rupture/erosion. Should more parameters be available as inputs and not need to be inferred (such as stain by use of Brillouin microscopy), results will accordingly be more precise. A major limitation of current computational models lies in the perceived difference of structural properties between plaque components in vivo and purified materials used to determine inserted parameters. Despite the above, advances in algorithms have led to reduction of necessary time to construct the model and obtain convergent solutions (i.e., functions describing plaque behavior) from weeks to less than 2 hours. The latter is comparable to offline time needed for 3‐dimensional reconstruction of IVUS and OCT data; thus, it may herald active application of models to clinical practice in the near future so as to evaluate the atherosclerotic plaque not as an isolated structure, but as part of a living vessel.
Although relatively scarce, some clinical data exist advocating the added prognostic value of integrating computational models in clinical practice. More specifically, even in the negative Prediction of Progression of Coronary Artery Disease and Clinical Outcome Using Vascular Profiling of Shear Stress and Wall Morphology (PREDICTION) study, wall shear stress was found predictive of a future decrease in luminal area and clinically significant obstruction, whereas balloon‐induced rupture was noted to occur at the site of the maximal structural stress in a 2001 study using IVUS to generate a finite‐element model. More important, in patients presenting with ACSs, increased stress values were noted in high‐risk areas, suggesting a potential cause‐and‐effect relation. Obviously, large, well‐designed prospective studies and, subsequently, randomized, clinical trials are currently lacking and sorely needed to verify or refute the validity of the above integrative approach.
_
There is ample evidence of a close relationship between plaque morphology and patient outcome. Although invasive modalities offer the highest spatial resolution, the best imaging technique is still to be determined. However, characterization of vulnerable plaque is not limited to morphologic assessment; molecular imaging can add significant relevant information concerning tissue inflammation and subclinical thrombosis as seen in the figure below.
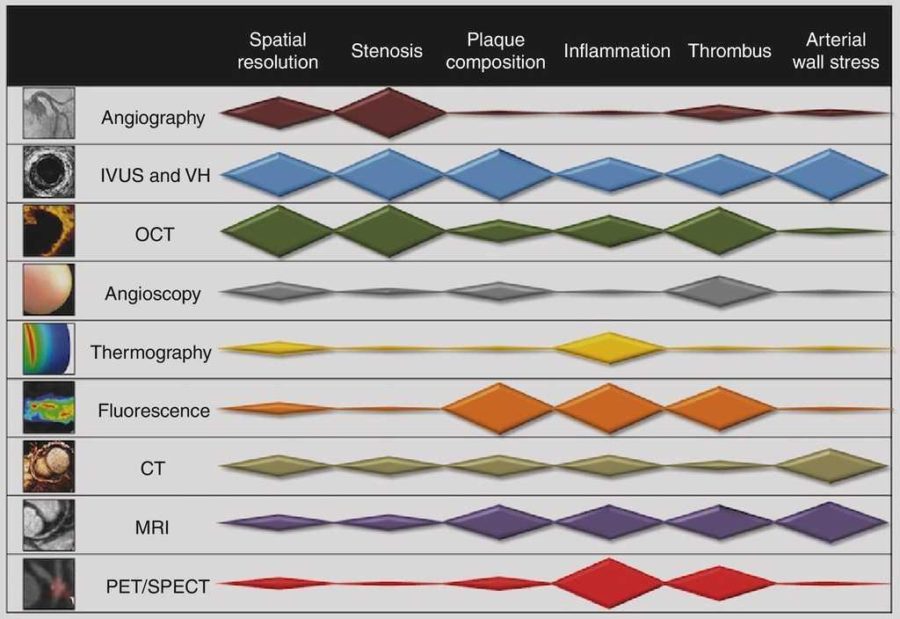
Figure above shows capabilities of cardiac imaging techniques for the assessment of different pathophysiological targets in vulnerable plaque. CT: computed tomography; IVUS: intravascular ultrasonography; MRI: magnetic resonance imaging; OCT: optical coherence tomography; PET: positron emission tomography; SPECT: single‐photon emission computed tomography; VH: virtual histology.
_______
_______
Morphological and biological imaging:
The morphologic characteristics of atherosclerotic plaques can be targeted by invasive and non‐invasive imaging modalities such as angiography, intravascular ultrasonography (IVUS), optical coherence tomography (OCT), computed tomography (CT) and magnetic resonance imaging (MRI). However, molecular imaging offers better discrimination of plaque components. To give an overall assessment of vulnerable plaque, imaging modalities need to target plaque morphology and structure, inflammation, apoptosis and thrombosis, and provide information on flow and wall stress.
Plaques with nearly similar morphology in terms of lipid core and fibrous cap may look similar with diagnostic imaging aimed at morphology only. However, they might look very different using diagnostic methods capable of detecting activity and physiology of the plaques.
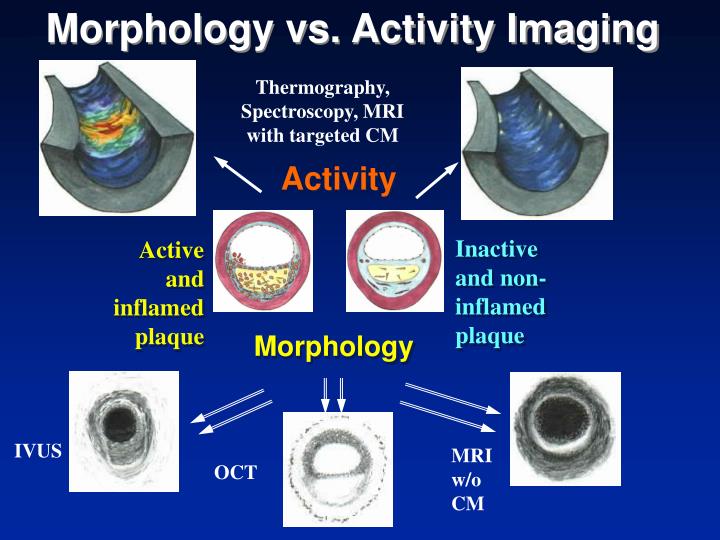
The top left plaque is hot (as evidenced in a thermography image), whereas the top right plaque is inactive and detected relatively as a cool plaque.
_
Among the anatomical imaging tools (Figure below, right), CT and MRI hold promise given their non-invasive character. Whereas MRI provides excellent soft tissue contrast and a better contrast resolution, CT allows much shorter scanning times. However, the hazard of exposing patients to radiation is a concern. The invasive approaches IVUS-VH and OCT/OFDI offer high-excellent spatial resolution. The combination of anatomical with biological imaging using hybrid techniques, i.e. PET–CT or PET–MRI, appears to be particularly attractive. Among those, targeted imaging that measures inflammation holds the greatest promise to identify vulnerable plaques by reporting on macrophages and other mechanisms involved in plaque vulnerability (Figure below, left) such as adhesion molecules, proteases, and matrix components.
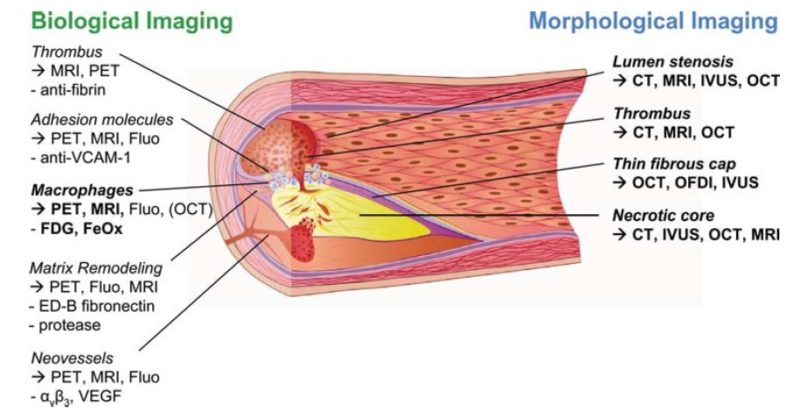
Figure above illustrates morphological and biological tools for visualizing vulnerable plaques. Modalities with clinical applications are given in bold. Optimal patient selection may be reached by integrating individual risk factors, clinical data, and key plasma biomarkers.
_
Targeting inflammation, apoptosis and thrombosis in the vulnerable plaque:
Traditionally, atherosclerosis was only diagnosed at advanced stages of the disease by documentation of stenosis or by perfusion assessment. However, new imaging modalities enable vessel wall composition to be assessed down to the cellular and molecular level. Plaque inflammation is strongly associated with plaque rupture and thrombosis, a hallmark of the ACS culprit lesion. Additionally, active and apoptotic leukocytes release enzymes which promote endothelial injury, cytolysis, and plaque disruption with subsequent thrombosis.
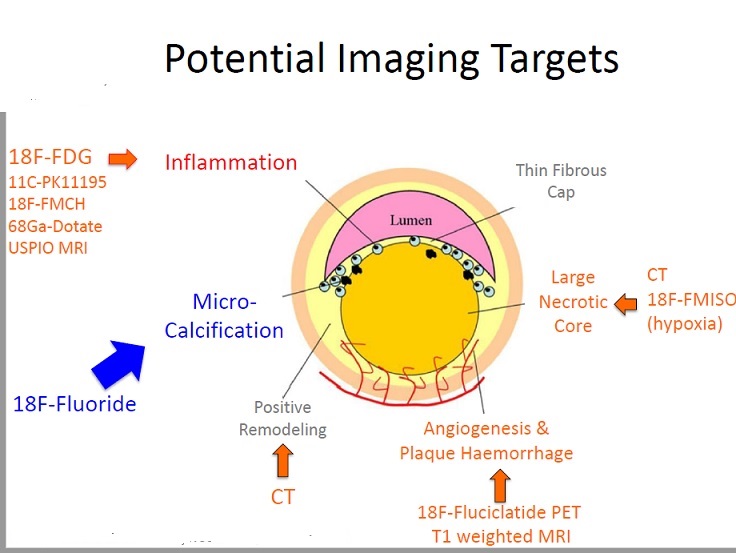
Figure above shows various invasive imaging modalities used to assess inflammation, thrombosis and apoptosis. They include thermography, OCT, angioscopy, intravascular photoacoustic (IVPA) imaging and IVUS.
______
______
Coronary angiography:
It is well established that, although an individual severe coronary stenosis may become acutely occluded, this is a more frequent occurrence in less obstructive plaques. Indeed, in approximately 70% of patients presenting with myocardial infarction, the lesions leading to occlusion have been reported as <50% stenotic. Although the coronary angiogram fails to identify most of the atherosclerotic changes affecting the vessel, it has provided some information about plaque vulnerability. In addition to identifying stenosis, the angiogram, particularly in patients with acute coronary syndromes, may reveal a complex, irregular interface between the luminal surface of the plaque and the injected contrast material. Angioscopic and autopsy studies have documented that these complex lesions are caused by plaque rupture, intra plaque hemorrhage, and intraluminal thrombosis. Complex lesions have a moderately increased likelihood (20–40%) of causing progression of stenosis within the year after their discovery, presumably because of organization of the initial thrombus followed by repeat rupture. Such lesions have also been linked to elevations in serum inflammatory markers and subsequent events. The finding of multiple complex lesions in an angiogram may be a sign that the patient (as opposed to the individual plaque) is vulnerable because of an active systemic inflammatory process that will produce numerous vulnerable plaques in the future. The coronary angiogram has also been used to identify patients with paradoxical coronary vasoconstriction, which has been linked to future cardiovascular events.
_
Intracoronary angioscopy:

Intracoronary angioscopy is an imaging technique that uses optical fibers to allow direct visualization of the plaque surface, the presence of thrombus and the color of the luminal surface. A normal artery appears as glistening white, whereas a plaque can be categorized based on its angioscopic color, such as yellow or white. Additionally, thrombus can be identified as white (platelet rich) or red (platelets and erythrocytes).
Atherosclerotic plaques of coronary arteries can be classified into two types according to color by coronary angioscopy. Yellow plaques are frequently observed at the site of a culprit lesion in the setting of acute coronary syndromes. These plaques have thin fibrous caps with lipid-rich cores and inadequate collagen content. In contrast, white plaques have thick fibrous caps or are completely fibrous. Various studies suggest that yellow plaques are more vulnerable to rupture than white plaques. Yellow plaques are softer and more distensible and are associated with compensatory enlargement more frequently than white plaques. Uchida and co-workers performed intracoronary angioscopy in 157 patients presenting with stable angina. In a 12-month follow-up period, ACS occurred more frequently in patients with glistening yellow plaques (69%) than in those with white plaques (3%).
Intracoronary angioscopy can also be applied as a tool for monitoring changes in plaque morphology following pharmaceutical therapy. Using this technique, Takano and colleagues were able to demonstrate an effect of preventive treatment with atorvastatin. Interestingly, lipid-lowering therapy with atorvastatin changed plaque color and morphology as determined by angioscopy, thereby suggesting plaque stabilization.
A major limitation of angioscopy remains that, as with OCT, the technique requires a blood-free field, while investigation is restricted to a limited part of the vessel.
_
Angiogram and corresponding angioscopic pictures:
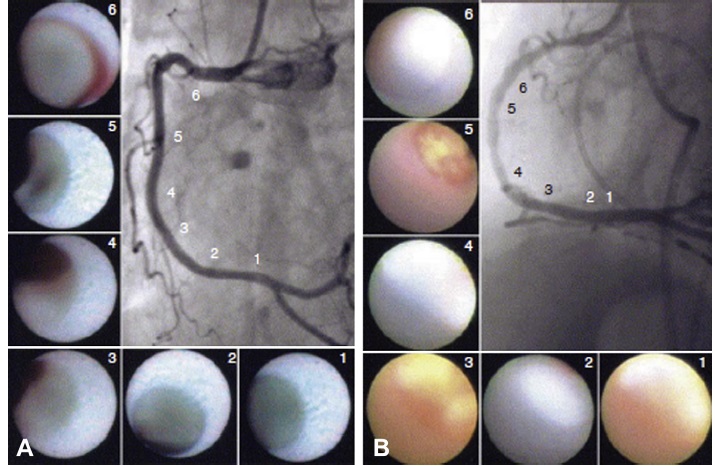
In figure above, Left panel shows normal angiogram and angioscopy. Right panel shows glistening yellow plaques at segments 5,3,1 and corresponding lesions on angiogram.
______
______
Physiological assessment: Fractional flow reserve (FFR):
Fractional flow reserve (FFR) is increasingly used to identify physiological important lesions which angiographically appear non-flow limiting. It is defined as a ratio of the pressure distal to a stenosis in relation to the pressure proximal to the stenosis under maximum hyperaemia, normally induced by either intra-coronary or peripheral adenosine infusion. A FFR of 0.75 means that a given stenosis causes a 25% drop in blood flow pressure which is considered as a significant stenosis. The DEFER investigators reported deferral of percutaneous coronary intervention in intermediate lesions with an FFR of > 0.75 is safe with a low risk of acute coronary syndrome presentations; <1% after 5 years of follow-up. The Fractional Flow Reserve Versus Angiography for Multivessel Evaluation (FAME) trial further demonstrated that the use of FFR in patients with multiple vessel coronary who are undergoing percutaneous coronary intervention with drug-eluting stents significantly reduces the mortality, nonfatal myocardial infarction and revascularization at 1 year compared with angiographically-guided group. In addition, Versteeg et al. demonstrated that the monocyte toll-like receptors 2 and 4, which are related to plaque vulnerability, were significantly higher in patients with FFR <0.75 than in patients with an FFR of >0.80. This suggests that plaque vulnerability may be preceded by ischaemia. To date, there is no clinical trial to date using FFR to detect vulnerable plaque per se, therefore, the role of FFR in the detection of vulnerable plaques and management of vulnerable patients requires further evaluation.
______
______
Intravascular ultrasound (IVUS) on the vulnerable plaque:
IVUS technology was invented in 1972. An ultrasound unit is mounted on the tip of a catheter. This unit can consist of a single element piezoelectric material with a frequency range between 30 and 45 MHz or of an array of 64 elements with a center frequency of 20 MHz. With regard to single element catheters, the beam will be steered by mechanically rotating the element, while for the array, the beam will be steered electronically. Rotation devices are able to produce an image with a resolution between 100 and 150 μm. Phased array catheters are generally easier to set up and more flexible, but historically have suffered from lower resolution when compared to their mechanical counterparts, and this may result in poorer near field imaging. These catheters can be advanced through the groin, then through the aorta all the way into the coronary arteries, thus a tomographic image of the vessel, vessel wall and atherosclerosis can be created.
In the normal artery, two interfaces are usually observed, one at the border between blood and the leading edge of the intima and a second at the external elastic membrane (EEM), which is located at the media adventitia border. The tunica media is relatively sonolucent and, in good quality images, it can be visualised as a distinct, relatively sonolucent layer. In 30% to 50% of normal coronary arteries, the thin intima reflects ultrasound poorly, so it is not visualised as a separate layer. Lumen measurements are performed using the interface between the lumen and the leading edge of the intima.
Greyscale IVUS helps us to potentially determine vessel lumen size, distribution and severity of atherosclerotic plaque. In addition, it might be particularly useful in detecting the cross sectional area. Lipid-laden lesions appear hypo echoic, fibro muscular lesions generate low-intensity or “soft” echoes, and fibrous or calcified lesions are relatively echogenic. Using IVUS Greyscale we can distinguish four kinds of plaques based on the acoustic signal, considering that the echogenicity and texture of different tissue components may exhibit comparable acoustic properties and therefore appear quite similar. Thus, IVUS is helpful at determining the echogenicity of vessel wall structures but it is not consistently able to provide actual histology. In a recent study by Low et al., the accuracy of Greyscale IVUS in defining plaque characteristics in vivo has been analysed, using OCT. The sensitivity for IVUS detection of lipid content was 24.1%, and specificity 93.9%. Although detection of calcification had the highest reproducibility, its specificity was only intermediate (66.4%) with an excellent sensitivity (92.9%). The detection of a thin cap (defined as <65 μm) was difficult since the current best resolution of IVUS is 100 μm. The sensitivity and specificity of IVUS for detection of intraplaque calcifications were 92.9% vs 66.4%, respectively, while for disruption were 66.7% vs 96.1%. The authors concluded that Conventional Greyscale IVUS may not be a reliable imaging modality for detection of lipid-rich and hence vulnerable plaques. However, Yamagishi et al. demonstrated the increased risk of rupture of an eccentric atheroma with a relatively large plaque burden, and a shallow echo lucent zone that was observed by Grey scale IVUS.
_
Virtual histology intravascular ultrasound:
Virtual histology intravascular ultrasound (VH-IVUS) can potentially differentiate plaque composition more accurately than conventional grayscale IVUS. Although conventional Grey scale IVUS improved the field of percutaneous coronary intervention (PCI), its ability to distinguish plaque sub-types is limited by inter- and intra-observer subjectivity as well as by its axial resolution. Virtual histology intravascular ultrasound (VH-IVUS) has moved the goal posts a little further by utilising similar principles to IVUS but allowing real-time quantification of plaque into differing subtypes. The technique is based on radiofrequency analysis of intravascular ultrasound backscatter signals. A combination of spectral parameters is used to develop statistical classification schemes for analysis of in vivo IVUS data in real time. Using these parameters, color-coded maps of plaque composition for each cross-sectional image are provided and are superimposed on the grayscale IVUS images. Statistical classification trees sorted the radiofrequency data based on the combination of spectral parameters into one of four plaque components: fibrous, fibro-fatty, necrotic core and dense calcium. The initial model performed well with predictive accuracy of 90.4%, 92.8%, 90.9% and 89.5% for fibrous, fibro-fatty, calcified and calcified necrotic, respectively.

_
_
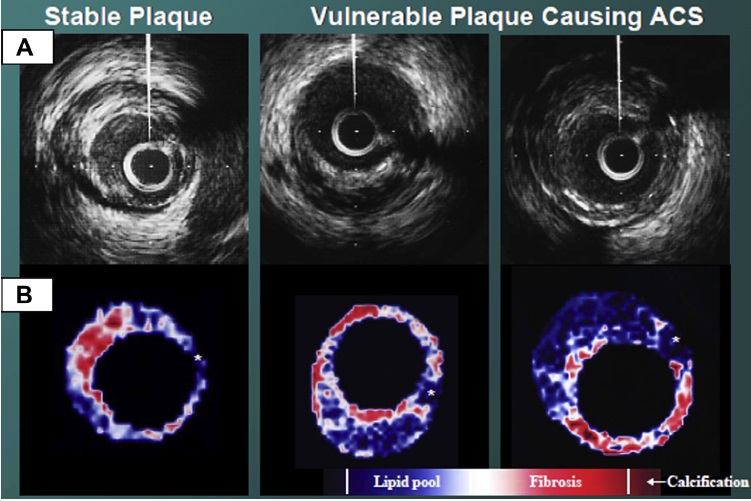
Figure above, A: Grey scale IVUS image of atherosclerotic plaque, B: Virtual histology image of the same: both showing thin cap with large core and heterogenous composition.
_
_
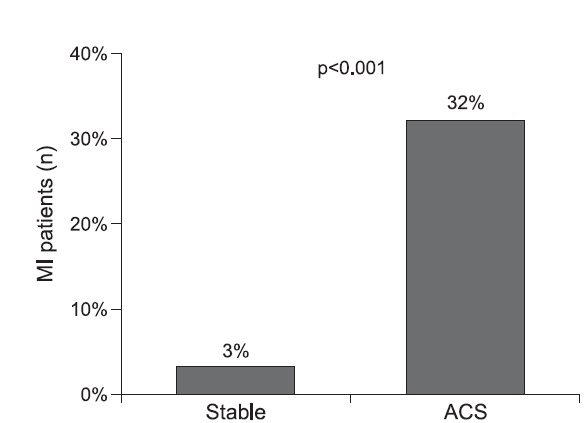
Figure above shows prevalence of thin-capped fibroatheroma (TCFA) in patients presenting with stable symptoms versus patients presenting with an acute coronary syndrome (ACS) evaluated by virtual histology intravascular ultrasound. TCFAs were more frequently observed in plaques of patients with ACS (32%) as compared to patients with stable symptoms (3%). The bar chart was constructed with data from Pundziute et al.
_
IVUS Elastography and Palpography:
The principles of elastography and palpography are based on the hypothesis that lipid-rich vulnerable plaques might have different mechanical propertied than stable, more fibrotic, plaques. Both are focused on the evaluation of local mechanical tissue properties, based on deformation of tissue. At a defined pressure, soft tissue components deform more than hard components. Consequently, in coronary arteries, regions with soft tissue components within the vessel wall will deform unequally during systole and diastole as the blood pressure (excitation force) varies throughout the heart cycle. The deformation of the plaque is assessed using ultrasound and images obtained at different pressure levels are compared to determine local tissue compression. The radial strain in the tissue is calculated by cross-correlation techniques on the radio-frequency signal and can be displayed as a color-coded image. This information is plotted as an additional image to the IVUS echogram A plaque with a high strain region at the surface with adjacent low strain regions is defined as a vulnerable plaque. In an in vitro study, it was demonstrated that sensitivity and specificity of elastography to detect vulnerable plaques are 88% and 89%, respectively. High correlation between the strain in caps and the amount of macrophages and an inverse relation between the amount of smooth muscle cells and strain was also observed. These results have been also verified in vivo, where it was shown that fatty plaques have an increased mean strain value and high-strain spots are associated with the presence of macrophages.
Based on the similar principle, IVUS palpography is assessing the local mechanical tissue properties of the luminal surface of the vessel wall. One strain value per angle is calculated and plotted as a color-coded contour at the lumen vessel boundary. It has been recently suggested that the distribution of the strain in the three-dimensional (3-D) geometry of an artery is an important tool to identify the presence of vulnerable spots.
In human coronary arteries, the number of highly deformable plaques has been correlated with both clinical presentation and levels of C-reactive protein (CRP). Limitation of this technique remains rhythm disturbances that may produce poor signal quality and motion artifacts that may also influence the performance of the method of the available techniques.
_
Contrast Enhanced Ultrasonography (CEUS):
Recent evidence suggests that Contrast Enhanced Ultrasonography (CEUS) can be used as a molecular imaging tool to target inflammation and visualize the associated neovascularization in a vulnerable plaque. CEUS medium consists of microbubbles of gas, enveloped by a shell of different substances (albumin, lipid, polymer, etc.). Gas microbubbles are strong reflectors of acoustic energy, thus increasing the return signal after tissue interrogation with ultrasound. Contrast microbubbles have a diameter of just a few microns (usually <5 micro meter) and have been shown to behave as a true intravascular tracer. CEUS has the ability to image intraplaque neovessels that usually originate from the vasa vasorum in the adventitia which are linked to plaque vulnerability. There have been multiple studies which have confirmed the utility of CEUS in the detection of intraplaque neovascularization with several clinical studies demonstrating that CEUS could help in the differentiation between stable and unstable plaques. Additionally, plaque vascularization measured by CEUS has been shown to correlate positively with 18F-fluorodeoxyglucose (FDG) uptake measured by PET/CT in humans. Although further prospective studies assessing the association of CEUS-detected neovascularization with future cardiovascular events are required, CEUS may represent a promising, safe, and widely available tool for detection of a vulnerable plaque.
______
______
Optical coherence tomography (OCT):

Optical coherence tomography (OCT) is a unique high-resolution imaging technique that uses low coherence, near infrared light for intravascular imaging of the coronary artery wall. It has an excellent spatial resolution of 10-20 μm, which is ten times higher than the resolution of IVUS. Furthermore, using histological controls, it has been demonstrated that OCT is superior to IVUS in detecting important features of vulnerable plaque components, including thickness of fibrous cap, thrombus and density of macrophages.
_
As shown in figure below, OCT visualizes 3 layer structures of normal coronary artery as well as plaque component of atherosclerotic lesions. The ability of OCT in tissue characterization of coronary atherosclerotic lesions has been well validated in clinicopathological studies. The drawback points of OCT are the relatively shallow depth of light penetration into the arterial wall in the comparison with ultrasound, and the necessity of blood removal by contrast medium during image acquisition.
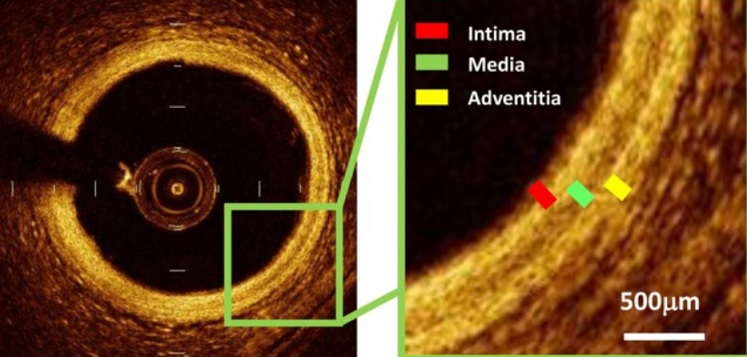
_
One of the first investigations to demonstrate the feasibility of plaque characterization with OCT in vivo was performed by Jang et al. Using this technique the authors reported a higher frequency of thin capped fibroatheroma in patients with ACS as compared to patients with stable angina pectoris. Kubo et al compared the assessment of culprit plaque morphology by OCT to grayscale IVUS and coronary angioscopy. The authors concluded that OCT was superior in identifying the thin-capped fibroatheroma and thrombus, and that OCT was the only modality that could distinguish the thickness of the fibrous cap.
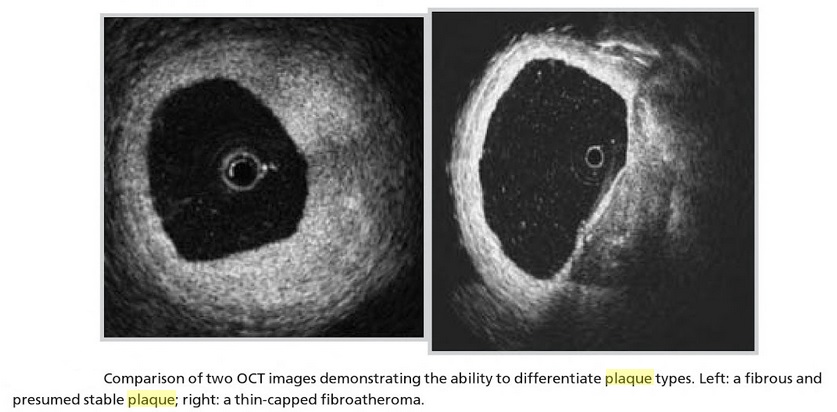
Another interesting feature of OCT is that it enables quantification of macrophages within fibrous caps. Tearney and colleagues showed in vitro, by comparing OCT images to histological specimens, that a high positive correlation exists between OCT measurements and fibrous cap macrophage density (r=0.84). In vivo, Raffel and colleagues demonstrated a significant relationship between systemic inflammation (white cell blood count) and macrophage density in fibrous caps identified by OCT.
OCT is the only imaging modality available for clinical use that is capable of measuring the fibrous cap thickness overlying a lipid plaque, therefore enabling the detection of TCFA. Kume et al demonstrated that after accounting for tissue shrinkage during histologic preparation, there is a good correlation between OCT and histologic examination in determining fibrous cap thickness (r = 0.9; P < .001). Importantly, fibrous cap thickness varies according to the clinical presentation, as shown by in vivo studies using OCT measurements. Patients with STEMI were found to have a considerably thinner fibrous cap in comparison with patients with non-STEMI and stable angina. Takarada et al demonstrated that statin therapy significantly increased the fibrous cap thickness in patients with hypercholesterolemia at 9-month follow-up. Recent OCT study showed that atorvastatin therapy at 20 mg compared with 5 mg provided a greater increase in fibrous cap thickness in coronary plaques of patients with unstable angina pectoris. However, the current methodology for determining fibrous cap thickness is based on manual individual measurements of arbitrary points (i.e., the thinnest regions determined by visual assessment), which might cause high variability and reduced accuracy. Besides, such one-dimensional analysis of fibrous cap thickness does not take into account the three-dimensional (3D) morphology of coronary artery disease, which largely limited the advancement of the clinical knowledge in this field.
Aiming at overcoming this important limitation of previous studies, a semiautomated method that allows comprehensive quantification of fibrous cap thickness and 3D visualization of its longitudinal and circumferential distribution along the vessel has been investigated. The method has been found to be highly accurate, yet more consistent than human experts. Moreover, Galon et al demonstrated that the novel OCT-based 3D quantification of the fibrous cap showed thinner fibrous cap thickness and larger areas of TCFA in nonculprit sites of STEMI compared with stable angina. Although the mechanisms of fibrous cap rupture remain unclear, it is possible that its mechanical stability may not only depend on a focal, thin point, but rather on the thickness of confluent regions of thin cap distributed along the plaque. Therefore, we need to further investigate in a prospective fashion whether this more comprehensive methodology to identify and quantify different fibrous cap thicknesses along the plaque may refine our ability to predict future plaque rupture and its devastating consequences.
At present, it is important to realize that there are some important limitations in the use of OCT. Blood leads to significant attenuation of the emitted infrared light, therefore regular saline flushes or balloon occlusion of the artery is necessary for adequate imaging. Consequently, data acquisition is time-consuming and is therefore limited to focal lesion exploration. Furthermore, the penetration depth of near infrared light is only 1-2 mm. As a result, OCT is not able to visualize the complete plaque and vessel wall, and quantitative measurements of plaque and/or lipid volume are currently not possible. Although OCT seems to be the most suitable imaging system due to its high resolution and unique ability to measure fibrous cap thickness, neovascularization, and inflammation, potential methodologic limitations observed in the majority of the studies that utilized OCT as reference might have precluded a better understanding of this complex subject. However, a second-generation OCT technology, namely optical frequency domain imaging (OFDI), has recently been developed to enable imaging of the coronary arteries with a short, non-occlusive saline flush and rapid spiral pullback.
The information acquired by OCT and IVUS are complimentary as OCT can produce high resolution images to help assess fibrous cap thickness and macrophage infiltrations of intact coronary atherosclerotic plaques while IVUS provides details of penetration and tissue characterization of the lesions.
______
______
Computed tomography (CT):
The possibility to non-invasively visualise coronary anatomy is the main reason for the current interest in CT. Its high temporal and spatial resolution allows detailed anatomical delineation of large and medium-sized vessels, so this technique can be considered the most accurate clinical method for non-invasive coronary angiography. In 2000, 4-slice CT systems allowed coronary artery imaging for the first time, but only the proximal parts of the coronary artery were interpretable because limited spatial and temporal resolution restricted their clinical value for coronary artery visualisation. With the introduction of 16- and 64-slice systems, improved temporal and spatial resolution, as well as substantially shorter scan times led to improved image quality throughout the entire coronary tree. A meta-analysis conducted in 2007 by Vanhoenacker et al. demonstrated a significant improvement in the accuracy for the detection of coronary artery stenosis for 64-slice CT when compared with previous scanner generations. The pooled sensitivity and specificity for detecting a greater than 50% stenosis per segment were 93% and 96% for 64-slice CT, 83% and 96% for 16-slice CT, 84% and 93% for 4-slice CT, respectively. The consistently high negative predictive value of CT angiography suggests that this exam should be recommended to rule out coronary stenoses. It may be also used to visualise earlier stages of coronary atherosclerosis. Coronary calcium, a surrogate marker for the presence and amount of coronary atherosclerotic plaque, can be detected and quantified by non-contrast CT, allowing us to stratify asymptomatic individuals with a stronger predictive power than stratification based on traditional risk factors. Coronary calcium measurements by CT may be useful in patients who, based on prior assessment of standard risk factors, seem to be at intermediate risk for future coronary artery disease (CAD) events. It may also be valuable when deciding to start lipid lowering therapy. Inoue et al. evaluated the plaque changes (plaque, low attenuation plaque and lumen volume, remodelling index) by serial coronary CT angiographies in 26 statin treated patients as compared to eight subjects who refused the statin treatment (controls). Serial CT angiographic evaluations of coronary plaques allowed for the assessment of statin treatment related reduction of the plaque and necrotic core volume. Although larger clinical studies are needed, a CT preliminary assessment of the plaque composition might be useful to evaluate antiatherosclerotic effects of statin treatment.
The high CT attenuation of calcified structures allows for a simple visual and qualitative characterisation of plaque composition by dividing coronary lesions into calcified, non-calcified and mixed plaques. Surprisingly, a higher prevalence of non-calcified and mixed plaques was found in the culprit lesions of patients with ACS, while patients with stable angina had more commonly calcified lesions. This evidence suggests that diffuse intraplaque calcification might be not associated with vulnerability.
The culprit lesions of patients with AMI, unstable angina and stable angina have been shown to have different calcification patterns. In particular, in patients with AMI spotty calcified deposits within an arc of ≤90° were found. On the basis of the good correlation between intravascular ultrasound (IVUS) and multidetection CT (MDCT) in the evaluation of coronary plaques, and particularly the excellent sensitivity of MDCT in differentiating calcified and soft components, MDCT can be considered as a promising non-invasive standard criterion to accompany the invasive imaging modalities.
_
Computed tomography for detection of vulnerable coronary plaque:
The majority of acute coronary syndromes occur without warning due to rupture of coronary atherosclerotic plaques. Coronary lesions prone to rupture reveal several high-risk features that can be identified before the development of devastating clinical events, using modern imaging modalities. Recent advances in coronary computed tomography angiography (CCTA) allow the identification, quantification, and risk stratification of coronary plaques. Unquestionably, attempts to identify high-risk plaques by CCTA provide a unique opportunity to implement targeted preventive measures and improve prognosis. Identification of plaque composition is based on measuring the attenuation coefficient which is displayed as a CT number relative to the attenuation of water (0 Hounsfield units HU) and air (-1000 HU). Calcifications appear as hyperdense, fibrous tissue as isodense and lipid core, intra-plaque hemorrhage and thrombus as hypodense. However, using the absolute CT numbers to define different plaque constituents, especially of soft plaques, is influenced by various factors that could limit its accurate assessment, rendering the exact definition of low attenuation plaque values with the currently available techniques more challenging. These factors include; attenuation of intracoronary contrast medium, tube voltage, degree of stenosis, use of different reconstruction filters.
_
The rationale for the identification of vulnerable plaque by coronary computed tomography angiography:
Histological features that have been identified to be associated with vulnerable plaque include a large necrotic core and a thin fibrous cap, plaque size and positive remodeling, plaque vascularisation, and macrophage infiltration. The limited spatial resolution of current CT technology (≈ 400 µm) precludes the direct visualisation of macrophage infiltration or the thickness of the fibrous cap. Similarly, although plaque vascularisation may theoretically be assessed by quantitation of vasa vasorum density using micro-computed tomography, this novel technology is still in its infancy and is currently unavailable for clinical use. For this reason, quantitative measurement of plaque size (defined by plaque burden and plaque remodeling) seems to be the most practical approach for the identification of potentially vulnerable plaques by CCTA. This can be further substantiated by the histopathological investigations showing that (1) the mean length of the TCFA’s necrotic core is 8 mm, and (2) the area of the necrotic core is most often > 1.0 mm2 – both of which are over the plaque detection threshold (> 1 mm plaque thickness) for current CT scanners. Another strategy for the detection of vulnerable plaque by CCTA includes qualitative assessment of large lipid pool of cholesterol-rich necrotic core as determined by low attenuation or napkin-ring sign. Moreover, the majority of TCFA occur in the proximal segments of the main coronary arteries corresponding to the largest vessel diameter where CCTA has the highest accuracy for plaque identification. Thus, both the quantitative and qualitative analysis of vulnerable plaques by CCTA seems technically justified and feasible.
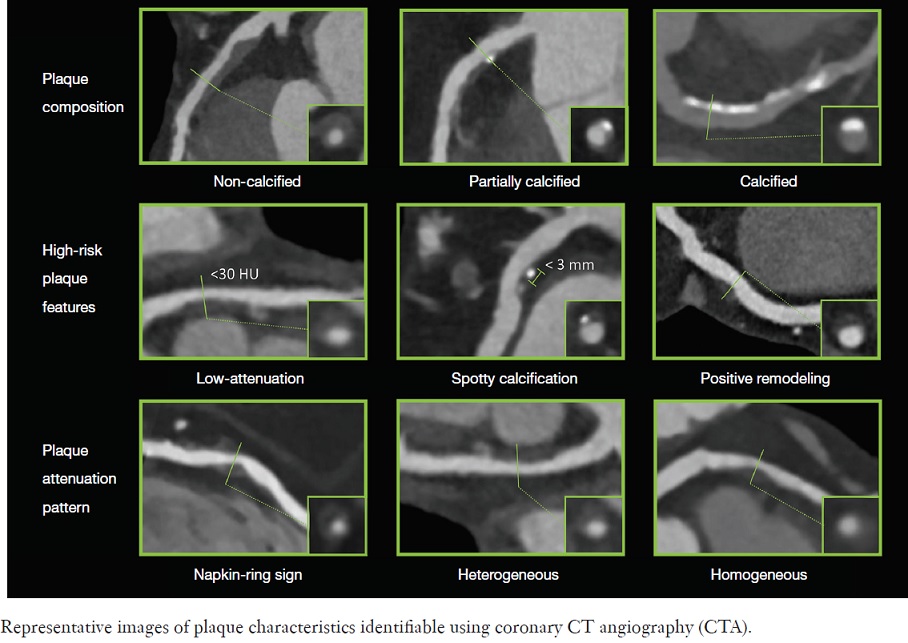
_
Multi-detector Computed Tomography:
The multislice CT (MSCT), or multi-detector- row CT (MDCT), is a CT system equipped with multiple rows of CT detectors to create images of multiple sections. First-generation scanners, or “conventional” CT, utilized a single X-ray source and single X-ray detector cell. Over the past two decades with the tremendous advances in technology, MDCT presented a breakthrough in cardiac CT imaging technology by 1) increasing the number of detector rows from 4 to 320 thus increasing the coverage in the Z-axis up to 16 cm in a single heart beat with a single gantry rotation, 2) speeding up the gantry rotation, enough to freeze the cardiac motion by capturing images within the relatively brief period of cardiac diastasis. The use of dual-source MDCT system has markedly improved the temporal resolution to approximately 85 msec, resulting in a shorter scan time and 3) decreasing the thickness of the detectors to 0.5-0.625 mm thus increasing the spatial resolution to image submillimeter structures. This has allowed more accurate and detailed imaging of coronary plaques morphology and composition.
MDCT has caught up with coronary angiography, showing an excellent diagnostic accuracy in diagnosis of obstructive CAD when compared to coronary angiography as the gold standard. In a recent meta-analysis of 188 studies (from 2004 to 2011), the mean sensitivity and specificity of MDCT were 97% and 87% respectively.
Role of MDCT in Detection of Vulnerable Plaque; Morphology and Composition:
The potential for MDCT evaluation of coronary plaques is enormous, given its noninvasive nature and its ability to evaluate the entire coronary arterial tree in contrast to IVUS.MDCT can help detect the suggested triad of the main features associated with plaque vulnerability namely; positive remodeling, low attenuation (<30 HU), and spotty calcifications. Other imaging phenomenon, that has been reported to be pathologically related to thin cap fibroatheromas the “napkin ring” sign, defined as the presence of a ring of high attenuation around plaque; with a higher CT attenuation than those of the adjacent plaque but no greater than 130 HU in order to differentiate from calcium depositions.
_
Detection of Vulnerable Plaque in Asymptomatic “Vulnerable” Subjects:
The current appropriateness criteria do not recommend using computed angiography as a screening tool in asymptomatic population considering the risk of radiation and contrast media, cost and lack of supporting evidence. Motoyama et al. studied 1059 asymptomatic subjects by MDCT where atherosclerotic plaques were analyzed for the presence of the three vulnerability features; the plaque characteristics of lesions resulting in ACS during the follow-up of 27 ± 10 months were evaluated. It was shown low attenuation and/or positive remodeling independently predicted ACS (hazard ratio=22.8, CI=6.9-75.2, p<0.001) with a significantly higher likelihood of ACS than 2 feature negative plaques or no plaques (p<0.001).
_
Coronary Artery Calcium (CAC) Score:
In June 2000, the American College of Cardiology (ACC) and American Heart Association (AHA) Consensus Panel wrote in the Journal of the American College of Cardiology: “Coronary calcium is part of the development of atherosclerosis; it occurs exclusively in atherosclerotic arteries and is absent in the normal vessel wall.” In the 1980’s US cardiologists lead by Dr. Arthur Agatston defined a method to assess the amount of coronary artery calcium by using electron beam computed tomography, otherwise known as ultrafast CT scan. The presence and extent of coronary calcium are first and foremost markers of the extent of atherosclerosis within the coronary arteries. Nonetheless, it is important to understand that the coronary calcium score does not necessarily reflect the severity of narrowing (the degree of stenosis). Still, a patient with a high calcium score is more likely to have a significant narrowing of a coronary artery than a patient with a low calcium score. An individual without coronary artery calcification is very unlikely to have a severe narrowing of a coronary artery. Although cardiovascular events can occur in patients with very low calcium scores, the incidence is very low. The amount of calcium in the walls of the coronary arteries, assessed by calculating the coronary calcium score, appears to be a better predictor of risk than standard risk factors.
Based on a number of studies, the following definitions are used to relate the coronary artery calcium score to the extent of atherosclerotic coronary artery disease:
Coronary calcium score 0: No identifiable plaque. Risk of coronary artery disease very low (<5%)
Coronary calcium score 1-10: Mild identifiable plaque. Risk of coronary artery disease low (<10%)
Coronary calcium score 11-100: Definite, at least mild atherosclerotic plaque. Mild or minimal coronary narrowing likely.
Coronary calcium score 101-400: Definite, at least moderate atherosclerotic plaque. Mild coronary artery disease highly likely. Significant narrowing possible
Coronary calcium score > 400: Extensive atherosclerotic plaque. High likelihood of at least one significant coronary narrowing.
Coronary artery calcium (CAC) is defined as a hyper-attenuating lesion >130 HU within an area of ≥ 3 pixels. Agatston score has been widely used to quantify CAC score by multiplying the lesion area (mm2) by a density factor (between 1 and 4).
_
The value of CAC (coronary artery calcium) scoring for risk stratification has been extensively studied. A large clinical trial by Greenland and colleagues showed the distinct incremental value of CAC scoring over the Framingham risk score in asymptomatic patients. In addition, Detrano et al demonstrated that CAC performed equally well among the four major racial and ethnic groups. In a even larger cohort of 25,253 asymptomatic individuals, Budoff and colleagues confirmed that CAC was an independent predictor of mortality and that risk scores increased proportionally with higher CAC scores. Particularly in patients initially classified as being at intermediate risk, knowledge of the extent of CAC may be valuable for refining risk stratification and determining further management.
_
Figure above shows coronary calcium on non contrast-enhanced multislice computed tomography (MSCT) axial images. Calcifications appear as bright white dense structures on MSCT. Panel A shows a 57-year-old patient without evidence of coronary calcifications in the left anterior descending coronary artery (LAD). Panel B shows a 53-year-old patient with calcifications in the LAD. AO – aorta.
_
In addition to risk stratification, it has been suggested that CAC scoring may allow noninvasive monitoring of changes in atherosclerotic plaque burden. Several investigations have demonstrated a halt in progression or even regression of coronary calcifications as a result of reductions in serum low-density lipoprotein (LDL) cholesterol concentrations. However, other investigations failed to show such an effect, despite effective reductions in systemic inflammation or LDL cholesterol concentrations. It is possible that changes in calcified plaque burden may not adequately reflect changes in total atherosclerotic plaque burden. Moreover, it has been suggested that plaque stabilization may even be associated with a relative increase of coronary calcifications rather than a decrease. Indeed, it is important to realize that the presence or absence of calcium itself is not a direct marker for vulnerability. Since no information is obtained about the presence of non-calcified plaques, CAC scoring does not allow for a reliable distinction between potentially unstable versus stable plaques.
_
Technical Limitations of CTCA:
Some technical factors might affect the role of CTCA in characterization of coronary plaques. These limitations include: 1) Radiation exposure; typical effective dose of radiation for retrospectively gated reconstruction is about 15 mSv, radiation doses delivered have markedly reduced to less than 2 mSv with the advent of newer prospective gating and ultra-low-dose protocols, these advances in technology are expected to further expand the CTCA applications and would permit follow-up studies to evaluate the progression of atherosclerotic plaques over time and with the use of interventional drugs like statins. 2) contrast agents; currently used contrast media in CTCA are considerably safe, however, there is a minimal risk that should be anticipated and dealt with in patients with contrast allergy or those with advanced renal insufficiency. 3) artifacts; various CT artifacts could limit the accuracy of CTCA in studying the plaque composition. Of note, dense calcification is a major limiting factor that could result in the blooming artifact phenomenon where calcium looks bigger than it actually is and 4) the significant overlap between tissue densities, rendering the accurate definition of plaque constituents more challenging.
__
Comprehensive plaque assessment by coronary CT angiography, a 2014 study:
Most acute coronary syndromes are caused by sudden luminal thrombosis due to atherosclerotic plaque rupture or erosion. Preventing such an event seems to be the only effective strategy to reduce mortality and morbidity of coronary heart disease. Coronary lesions prone to rupture have a distinct morphology compared with stable plaques, and provide a unique opportunity for noninvasive imaging to identify vulnerable plaques before they lead to clinical events. The submillimeter spatial resolution and excellent image quality of modern computed tomography (CT) scanners allow coronary atherosclerotic lesions to be detected, characterized, and quantified. Large plaque volume, low CT attenuation, napkin-ring sign, positive remodelling, and spotty calcification are all associated with a high risk of acute cardiovascular events in patients. Computation fluid dynamics allow the calculation of lesion-specific endothelial shear stress and fractional flow reserve, which add functional information to plaque assessment using CT. The combination of morphologic and functional characteristics of coronary plaques might enable noninvasive detection of vulnerable plaques in the future.
__
Non-invasive detection of coronary inflammation using computed tomography and prediction of residual cardiovascular risk (the CRISP CT study): a post-hoc analysis of prospective outcome data, 2018:
Coronary artery inflammation inhibits adipogenesis in adjacent perivascular fat. A novel imaging biomarker—the perivascular fat attenuation index (FAI)—captures coronary inflammation by mapping spatial changes of perivascular fat attenuation on coronary computed tomography angiography (CTA). However, the ability of the perivascular FAI to predict clinical outcomes is unknown.
This study shows that the perivascular FAI enhances cardiac risk prediction and restratification over and above current state-of-the-art assessment in coronary CTA by providing a quantitative measure of coronary inflammation. High perivascular FAI values (cutoff ≥–70·1 HU) are an indicator of increased cardiac mortality and, therefore, could guide early targeted primary prevention and intensive secondary prevention in patients.
______
______
Magnetic Resonance Imaging of Coronary Arteries and Plaques:
Cardiac magnetic resonance (CMR) offers the promise of radiation-free imaging of the coronary arteries, providing information with respect to luminal stenosis, plaque burden, high-risk plaque characteristics, and disease activity. In combination, this would provide a comprehensive, individualized assessment of coronary atherosclerosis that could be used to improve patient risk stratification and to guide treatment. However, the technical challenges involved with delivering upon this promise are considerable, requiring sophisticated approaches to both data acquisition and post-processing.
Histological studies have indicated that culprit plaques responsible for MI have certain pathological characteristics, including a large necrotic core, positive remodeling, a thin fibrous cap, angiogenesis, intraplaque hemorrhage, inflammation, and microcalcification (see figure below). Each of these represents a potential imaging target that might be used to refine our ability to identify high-risk coronary lesions. To better predict an individual person’s risk, it will be important to measure not just the plaque burden but also the high-risk plaque burden.
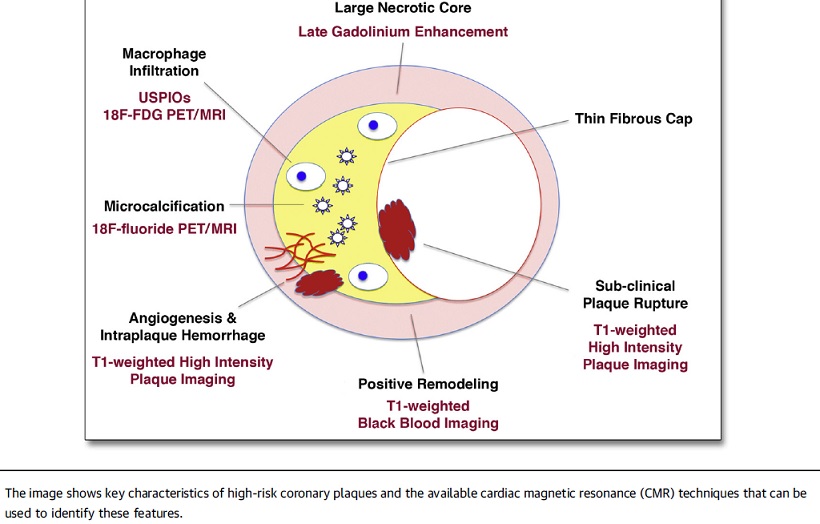
CMR has the potential to provide a multiparametric assessment of coronary atherosclerosis incorporating key information related to plaque burden, high-risk plaque characteristics, and disease activity alongside more conventional assessments of luminal stenosis. Considerable technical challenges remain in reliably translating these techniques in to the coronary arteries; however, if these can be met, then CMR holds real promise in improving our understanding of CAD, identifying patients at increased cardiovascular risk, and in delivering the promise of truly personalized health care for CAD.
_
High-intensity plaques on MRI as a novel surrogate marker of vulnerable coronary lesions:
Recently, high-resolution magnetic resonance imaging (MRI) has emerged as the leading non-invasive in vivo imaging modality for the characterization of atherosclerotic plaques. MRI has the potential to differentiate plaque components on the basis of biophysical and biochemical parameters. Moreover, MRI does not involve ionizing radiation and can be repeated sequentially over time. Oshita et al. reported in 2017 that coronary high-intensity plaques (HIPs) on non-contrast T1-weighted imaging (T1WI) were associated with the presence of high-grade yellow plaque with thrombus. This MR technique has been established for carotid imaging and can reliably identify carotid plaque with intraplaque hemorrhage. A study by Kawasaki et al. reported the observation of coronary HIP with non-contrast T1W cardiac magnetic resonance and investigated their relationship with multi-detector computed tomography, IVUS, and coronary flow by X-ray angiography. HIP was more likely to demonstrate positive remodeling, ultrasound attenuation, lower Hounsfield units, and transient slow flow after percutaneous coronary intervention (PCI). All these features have been associated with vulnerable plaques. Moreover, Noguchi et al. reported results from a single-center study investigating the usefulness of non-contrast T1WI for the detection of coronary HIP and its prognostic value. They demonstrated that the presence of HIP with a plaque-to-myocardial signal intensity ratio (PMR) of ≥1.4 was significantly associated with future coronary events in patients with stable coronary artery disease. In addition, Asaumi et al. and Hoshi et al. showed that coronary HIPs with high PMR are associated with periprocedural myocardial injury during PCI. Taken together, coronary HIP on non-contrast T1WI may have the potential for detecting high-risk plaque.
_
MRI is likely the most promising technique for cardiovascular imaging, since it does not require ionizing radiation, has a high spatial resolution and is non- invasive. Cardiac MRI (CMRI) is performed through techniques and principles that are used in body MRI. Current applications of CMRI are to detect and monitor the development of atherosclerotic plaques in peripheral vessels. The limitations encountered with CMRI are largely due to motion artifacts by both respiratory and cardiac movement. The size and tortuosity of the coronary vasculature has limited the use of CMRI especially when compared to the higher-quality images obtained of large vessel studies such as the aorta and carotid arteries. Several modifications, including the ‘‘black-blood’’ and free breathing techniques, have been implemented to enhance the quality of CMRI. MRI with contrast is, however, enhancing the characterization of the atherosclerotic plaque. Recent advances that allow image acquisition of multiple vascular beds, full body MRI, during a single procedure will further enhance the screening ability of MRI to detect atherosclerosis. Finally, newer 3 Tesla MRI scanners are expected to provide increased signal-to-noise ratio for better images and improve image resolution and speed the scanning time.
_
Studies have shown that MRI can detect features of the plaque associated with its vulnerability, including the lipid-rich necrotic core and the thin fibrous cap. An intact fibrous cap is generally seen as a continuous hypointense band against the bright lumen with a smooth surface during T1-weighted imaging, whereas an irregular surface or discontinuity of the hypointense band indicates recent hemorrhage and plaque rupture. Contrast-enhanced MRI (CE-MRI) is typically used to perform magnetic resonance angiography (CE-MRA) and can be supplemented with time-resolved angiography, flow measurement, vessel wall imaging, and plaque characterization for a more comprehensive assessment of vascular diseases.
_
Molecular MRI of Vulnerable Plaques:
Molecular MRI with targeted contrast agents contributes to a significant improvement in visualizing the characteristics of vulnerable plaques in smaller arteries. Nanoparticle platforms which harbor both (a) molecular probes with affinity for plaques or antibodies directed against plaque antigens, and (b) a reasonable payload of Gd as a T1 shortening agent have been used in several experimental studies. Inflammation is an important feature of atherosclerosis, and macrophages play a key role in the atherosclerotic process. Since the instability in an atherosclerotic lesion is promoted by the activation of mononuclear phagocytes, two MRI strategies have been used to detect this macrophage infiltration. The first technique uses gadolinium to detect the kinetics within the tissue that relate to phagocyte activation and mobilization. The other uses ultrasmall super paramagnetic particles of iron oxide (USPIO) to target macrophages in vivo. USPIO particles can be taken up by activated macrophages in vulnerable plaques and are visible on T2-weighted sequences as low signal intensity areas. The ATHEROMA trial was conducted to test the ability of USPIO to detect plaques in forty asymptomatic patients, demonstrating a significant reduction in plaque uptake with high-dose statin over a 3-month period. Although this trial did not show any significant association between USPIO signal intensity changes and subsequent cardiovascular and cerebrovascular events, it did show that USPIO was an effective method at detecting minute-to-minute changes in the cellular kinetics that were responsible for converting a stable plaque into an unstable one. A recent study has shown that increased vascular permeability using an MR albumin-binding contrast agent and T1-mapping served as a surrogate measure of plaque progression and instability, which has the potential to help stratify atherosclerotic disease progression. Newer contrast agents are being investigated which will further enhancement the ability of CMRI to delineate and identify plaque components. Gadofluorine has been shown to have high affinity for lipid-rich plaques and enhance MRI detection of soft plaques in experimental studies.
_
Intravascular MRI:
The non-invasive use of MRI to identify lipid in coronary plaques is limited by the small size of the target and cardiac motion. Invasive use of MRI in line with IVUS have been attempted. The feasibility of accurate in vivo imaging has been demonstrated in rabbits and swine. This approach showed significant differences in apparent diffusion coefficients between fibrous tissue, fatty streak, and lipid-rich necrotic cores.
_______
_______
Molecular Imaging of Vulnerable Plaque:
Molecular imaging is a type of medical imaging that provides detailed pictures of what is happening inside the body at the molecular and cellular level. Where other diagnostic imaging procedures—such as x-rays, computed tomography (CT) and ultrasound—offer pictures of physical structure, molecular imaging allows physicians to see how the body is functioning and to measure its chemical and biological processes. Molecular imaging includes the field of nuclear medicine, which uses very small amounts of radioactive materials (radiopharmaceuticals) to diagnose and treat disease. In nuclear medicine imaging, the radiopharmaceuticals are detected by special types of cameras that work with computers to provide very precise pictures of the area of the body being imaged.
Molecular imaging provides multiple imaging techniques to identify characteristics of vulnerable plaque including I) Inflammatory cells (the presence and metabolic activity of macrophages), II) synthesis of lipid and fatty acid in the plaque, III) the presence of hypoxia in severely inflamed lesions, IV) expression of factors stimulating angiogenesis, V) expression of protease enzymes in the lesion, VI) development of microthrombi in late-phase lesions, VII) apoptosis, and VIII) microcalcification.
Figure below shows Molecular imaging targets for the vulnerable plaque:
_
Imaging modalities of molecular imaging:
There are many different modalities that can be used for noninvasive molecular imaging. Each have their different strengths and weaknesses and some are more adept at imaging multiple targets than others.
Using dedicated tracers, nuclear imaging techniques such as single photon emission tomography (SPECT) and positron emission tomography (PET) can target distinct mediators and regulators involved in the cascade of atherosclerosis. As a result of increasing knowledge regarding the pathophysiology of atherosclerosis, several radionuclide-labeled tracers that serve as markers of inflammation, angiogenesis, apoptosis and lipid metabolism have been developed for plaque imaging. The rationale for the use of nuclear techniques in atherosclerosis imaging is that, thanks to its emission of gamma rays, a radiotracer can be co-localised with a cell or receptor of interest in the plaque, thus showing its functional characteristics. Those characteristics can be applied to non-invasively detect inflamed and potentially rupture-prone lesions. The main advantage of nuclear imaging is its excellent sensitivity which allows us to detect sparse targets in the nanomolar range using a low tracer dose. Even though single photon emission computed tomography (SPECT) and positron emission tomography (PET) were historically burdened by the low spatial resolution, hybrid imaging techniques have recently been introduced, adding the information of a molecular signal to the anatomical data obtained by CT and MRI. This progress has led to an increase in interest in the application of these techniques to the cardiovascular system.
_
Positron Emission Tomography (PET)
The underlying principle of nuclear imaging techniques, such as PET, is the use of a radiotracer, which emits gamma rays that can be localized to a cell or receptor in an inflamed plaque. This allows for noninvasive detection of an inflamed and vulnerable plaque prone to rupture in the near future. Following the discovery of fluorine-18-flurodeoxyglucose (FDG), a radiotracer which has increased uptake in carotid plaques in patients with ischemic stroke, several studies have found an association between FDG uptake and vulnerable plaques. Studies have proven that the amount of FDG uptake is directly proportional to the amount of inflammation in the plaque and that the use of statins reduces the inflammation as determined by the FDG uptake over time.
Although FDG-PET appears to provide a promising approach for the detection of inflammation in a vulnerable plaque, it does possess certain limitations. First, there are only a limited number of small prospective studies that have shown a correlation between adverse cardiovascular outcomes and increased FDG uptake in atherosclerotic plaque. Second, FDG uptake in a plaque can be influenced by mechanisms other than inflammation such as hypoxia, which may give false positive results. Therefore, even though FDG-PET seems to reliably detect an inflammatory plaque, it cannot currently be used as a predictor of outcome in a given atherosclerotic lesion.
_
Demeure et al. investigated both carotid inflammation and angiogenesis using FDG PET and contrast-enhanced ultrasound respectively, performed in patients undergoing carotid endarterectomy (11 were symptomatic and 19 asymptomatic). The contrast-enhanced ultrasound used a contrast agent that remains intravascular and so informs about new vessel formation rather than vessel leakiness. Imaging data were then compared with histological analysis of the excised carotid atheroma. This provided both validation of the imaging findings and an alternative assessment of the presence of inflammation and angiogenesis in the different plaque types. A good correlation between FDG uptake and macrophage burden was observed as previously reported. Similarly patients thought to have angiogenesis on ultrasound had a higher vessel density on histology than those that did not, although the binary nature of this assessment does represent a limitation of this technique.
The key finding of the study was that inflammation (assessed using both FDG PET and macrophage burden on histology) was increased in symptomatic versus asymptomatic patients despite similar plaque burden. This supports previous data. The same however was not true of angiogenesis, whether assessed by imaging or histology. This suggests that inflammation and angiogenesis may not be as closely related as previously thought and that angiogenesis may not be such a high-risk marker, at least in the absence of associated plaque haemorrhage.
_
The limitations of FDG-PET in vulnerable plaque imaging require us to focus on novel detectable markers of the plaque biology. In fact, depending on the radiotracer, a wide variety of physiological and pathological processes can be studied at the molecular level using PET. In 2006, Matter et al. observed a choline accumulation in the macrophages of activated atherosclerotic plaque in a murine model using Fluorine-18-labelled fluorocholine (18F-FCH) PET imaging. This technique allowed for better identification of plaques as compared to FDG-PET. In 2008, for the first time, Bacerius et al. described the use of this radiotracer in human abdominal aorta and common iliac arteries. Fluorine-18-labelled fluoromethylcholine (18F-FMCH) showed promising results in detecting non-calcified lesions of the vessel wall, both in solely structural wall artery and in combined lesions with structural wall artery and inherent calcified parts. On the other hand, solely calcified vessel lesions did not uptake the tracer at all. Another prospective study has been carried out recently in this field, analysing the 18F-NaF uptake in the coronary arteries by PET/CT. In this study, Dweck et al. stated that coronary uptake of 18F-NaF reflects active calcification in the atherosclerotic plaque, offering additional and complementary information to the CAC scoring detected by CT. Several prospective studies are now needed to determine the relationship between 18F-NaF uptake, morphological plaque characteristics and future cardiovascular events, and to identify novel detectable markers.
_
Single Positron Emission Computed Tomography (SPECT):
Both SPECT and PET work on the similar principle of radiofrequency signal uptake, but SPECT uses a different radiotracer which may be Iodine-123, Indium-111, or Technetium-99. SPECT differs from PET in several ways, including spatial resolution and imaging sensitivity. Oxidized LDL labelled to Technetium-99 has been shown to have the greatest sensitivity in detecting vulnerable plaques. Lecithin-like oxidized LDL receptor 1 (LOX-1) is a cell surface receptor for oxidized LDL that has been implicated in plaque instability and 99mTc-labeled anti-LOX 1 monoclonal IgG has been shown to have increasing accumulation in a vulnerable plaque, both of which can be detected by SPECT. Another proposed mechanism for the detection of plaque instability with use of SPECT imaging is the detection of apoptotic cells in a plaque with Annexin A5 as the marker for apoptosis. Annexin A5 is a protein which targets the phosphatidylserine surface expression of cells (such as macrophages and platelets) during the apoptotic process. 99mTc-labelled Annexin A5 has been shown to have increased uptake in an inflamed atherosclerotic plaque. A significant reduction in Tc-99m annexin A5 uptake after dietary modification and simvastatin therapy was observed by Hartung et al. Despite these promising results, the use of SPECT in the detection of inflammation or plaque instability is limited due to the lack of resolution and low specificity. Additionally, this technique is not cost-effective.
_
Matrix metalloproteinases (MMP) are released by activated macrophages and are therefore used to identify proteolytic activity in atherosclerotic lesions. MMPs modulate the degrading of the extracellular matrix and the thin fibrous cap of an atherosclerotic lesion, contributing to the vulnerability of the plaque. The feasibility of in vivo imaging of MMP activity using radionuclide-labeled MMP inhibitors has been shown in several animal models.
______
Hybrid techniques:
Several imaging platforms are currently available for targeted vascular imaging to acquire information on both anatomy and pathophysiology in the same imaging session using hybrid technology, such as PET/CT or PET/MRI. Indeed, PET imaging has relatively low spatial resolution, mandating the use of concurrent structural imaging (CT or MRI) to guide localization of the FDG signal, revealing whether the plaque is actually metabolically active or not.
Hybrid PET/CT enables measurement of functional and structural features of atherosclerosis, in which FDG uptake detects plaques with an ongoing inflammatory process, whereas CT identifies chronic calcification as a manifestation of the late stage of the diseases.
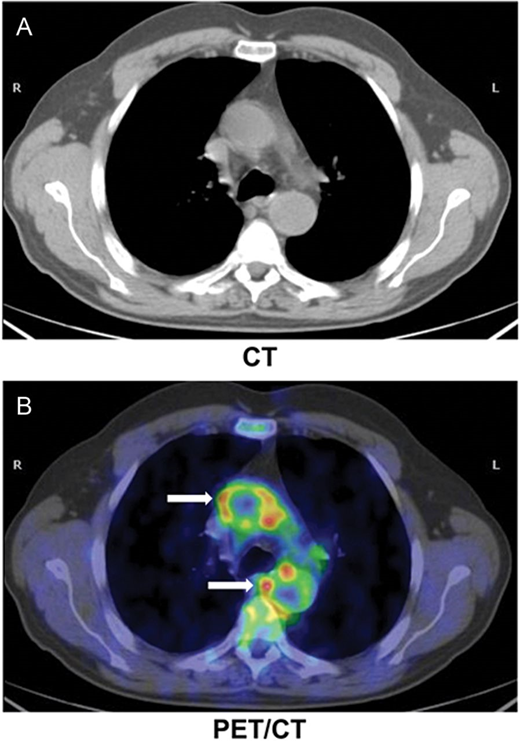
Figure above shows plaque detection by hybrid PET/CT. Contrast-enhanced middle-chest axial CT view shows parietal thin abnormality (thrombosis) of the ascending and descending aorta (A). Hybrid PET/CT scan demonstrates areas of inhomogeneous parietal FDG uptake in the thoracic aorta (arrows), suggesting an ongoing inflammatory process (B).
FDG-PET/CT has also been used to detect the anti-inflammatory effect of short-term statin treatment on aortic lesions, in correlation with the circulating inflammatory biomarkers. In particular, a significant reduction of FDG activity was reached after atorvastatin treatment. These findings provide in vivo evidence that the medium dose of atorvastatin (40 mg) might have a beneficial effect on plaque stability in 12 weeks, suggesting that a relatively low dose and a short treatment interval are sufficient to observe changes in plaque burden. Ishii et al. prospectively assessed the effect of 6-month therapy with 5 vs. 20 mg of atorvastatin on reducing FDG uptake in aortic plaques with PET/CT, and showed that only atorvastatin 20 mg significantly reduced plaque inflammation.
Experts agree no single modality has emerged as optimal for imaging vulnerable plaque. In fact, it may be a hybrid approach that ultimately meets the goal of imaging vulnerable plaque. The combination of IVUS, NIRS, OCT, fluorescence imaging and photoacoustic tomography technology in hybrid catheters will provide new synergistic information. Patel and his colleagues have reviewed IVUS, OCT and hybrid OCT-IVUS images of autopsy vessels with cardiologists and asked them to identify lipid, vulnerable plaque and calcium. They found cardiologists had a more accurate assessment of plaque when they viewed the hybrid images. This hybrid approach may address other challenges as well. Speed can be an issue with an IVUS catheter because it’s pulled back slowly, and the physician may be unsure of where he saw the lesion. An OCT catheter tends to be much faster, but localization can be difficult, so there may be multiple passes with a catheter which can result in harm. A single catheter with the advantages of both OCT and IVUS that localizes the lesion and the catheter may provide a more optimal alternative.
Emerging hybrid PET/MRI has considerable potential for cardiovascular imaging. In a recent study, the relationship between inflammation in PET and morphological features of carotid plaques associated with high risk of clinical events, such as lipid deposition, remodelling, and luminal irregularities, was studied in patients with PET and MRI co-registration. Carotid inflammation increased with the number of high-risk morphological features observed, supporting the concept that plaque inflammation and composition are closely linked. In the future, with new hardware developments and the introduction of combined PET and MRI scanners, it would be possible to add the most attractive aspects of both modalities in order to have a more profound look into the biology of atherosclerotic plaques.
_____
_____
There are specific advantages and disadvantages associated with each modality for imaging vulnerable atherosclerotic plaque in the coronary arteries. Certain technical and patient security challenges need to be overcome by molecular imaging modalities before they can become clinical tools. These imaging techniques should be tested in clinical trials to assess their real‐world prediction of coronary events; they are promising but, ultimately, the ability to identify patients who will benefit from intensive medical therapy, local intravascular therapy or preventive coronary bypass surgery will determine their success.
_
Multimodal imaging integrating coronary morphology, wall composition, characterization and wall shear stress can provide a highly accurate and predictive assessment of vulnerable plaque. For determining this integrated individual risk, invasive imaging modalities currently present various advantages over PET, MRI and CT; they provide greater anatomical detail, have tissue characterization capabilities and, particularly IVUS, assess inflammation and thrombosis using molecular imaging in addition to localizing areas of high wall stress.
In conclusion, much of the impetus for the development of new imaging modalities has come from basic science. In order to predict individual cardiovascular risk and to develop pharmacologic and interventional strategies to reduce acute coronary events, integrative clinical translation is crucial to the future success of vulnerable plaque imaging.
_______
Imaging Vulnerable Plaque: A Medical Necessity or a Scientific Curiosity? A 2017 study:
The potentially devastating nature of acute cardiovascular and cerebrovascular events makes identification of vulnerable plaques an important clinical goal. Investigators have fairly complete knowledge of plaque characteristics that determine plaque instability and include large plaque and necrotic core volumes, positive vascular remodeling, thin fibrous caps, and substantial cap and core inflammation. Although morphological features such as the plaque and core volumes and segmental remodeling can be identified by noninvasive imaging computed tomography (CT) angiography, fibrous cap thickness requires invasive assessment best undertaken with optical coherence tomography. The inflammatory component requires molecular imaging using single-photon or positron emission tomography (PET). Several radionuclide imaging approaches have been described to meet this clinical challenge, including 18fluorodeoxyglucose (18F-FDG) (a marker of glucose consumption by activated macrophages); 99mtechnetium-annexin V (a marker of apoptotic, predominantly macrophage, cell death), 18F-fluoromisonidazole (a marker of hypoxia in the necrotic core), F-18 sodium fluoride (a marker of microcalcification in the necrotic core), and 18F-galacto RGD (a marker of angiogenesis). Although 18F-FDG characterizes the inflammatory state within the plaques, more specific targets have been identified with variable success. For instance, radiolabeled mannose not only affirms the metabolic state of the inflammatory cells but also allows additional targeting of the M2 receptors, hence improving the targeting efficiency and target-to-background ratio.
Several investigators have described macrophage expression of type 2 somatostatin receptors as an alternative indicator of activated macrophages in the lesion. When monocytes enter tissue and transform into tissue macrophages, they express type 2 somatostatin receptors. A radiotracer developed for identification of neuroendocrine tumors, gallium-68 (68Ga)−labeled DOTATATE, localizes with nanomolar affinity in type 2 somatostatin receptors. Tarkin et al. compared 68Ga-DOTATATE localization in atheroma with FDG as a marker of increased macrophage metabolism. The investigators studied 42 patients, 18 with stable cardiovascular disease (CVD) and 24 with unstable CVD (10 with acute coronary syndrome and 14 with transient ischemic attack/stroke). The images obtained with FDG demonstrated uptake both in culprit and nonculprit lesions, whereas images recorded with 68Ga-DOTATATE had a higher concentration of tracer in the culprit vessel than in the nonculprit vessels. The 68Ga PET-CT images in the study depicted intense focal uptake in the culprit sites, suggesting that the studies should be readily interpretable.
Some of the investigators who contributed to the Tarkin et al. study participated in a previous investigation by Joshi et al., who compared NaF with FDG in 40 patients with stable angina and 40 patients with acute coronary syndromes, and reported NaF to be superior to FDG. Although more data are necessary, it is apparent that FDG imaging of atheroma does not differentiate between culprit and nonculprit lesions. The present enthusiasm for NaF and 68Ga-DOTATATE is based on small sample sizes. Both NaF and 68Ga-DOTATATE appear to work well, but by different mechanisms. NaF appears to localize in the necrotic core of atheroma that form microcalcifications and have sufficient inflammation to cause a local increase in vascularity. In contrast, 68Ga-DOTATATE localizes in receptors expressed by tissue macrophages, a cell type that is present in increasing numbers as lesions become more inflamed. To determine if NaF or 68Ga-DOTATATE is superior, it is important to have a multicenter prospective study of these agents in patients with CVD or cerebrovascular disease.
All eligible tests, whether noninvasive or invasive, have demonstrated relatively superior negative predictive value, but the positive predictive values remains too low to warrant a preemptive intervention. Consider the example of the landmark PROSPECT (Providing Regional Observations to Study Predictors of Events in the Coronary Tree) study in which 3 determinants of plaque vulnerability (i.e., thin cap, large plaque burden, and significant luminal stenosis) were described. The negative predictive value for a recurrent event arising from a nonculprit lesion was 99.7% in the absence of all 3 determinants. In contrast, the positive predictive value in the presence of all 3 determinants was slightly >18%. More importantly, only 1% of the nonculprit lesions displayed all 3 determinants, which brought the percentage of eventful lesions down to <0.2%. Authors long-term CT angiography study, in a large cohort of patients, demonstrated that the vulnerable lesions had to continue to gather larger necrotic core volumes over time to sustain plaque instability. Progressively enlarging necrotic cores are also likely to demonstrate persistent inflammation. Therefore, a consideration in the search for the best marker of vulnerable plaque would be to determine whether a vulnerable lesion evolves over time as a vulnerable lesion or stabilizes with time. In a serial intravascular study, in which 80% of the lesions were identified as vulnerable at baseline had stabilized over the ensuing year, a small proportion but equinumeric stable plaques progressed to instability. Thus, either the exploratory techniques need to define the serial behavior of the vulnerable lesions over time, or we may need to contend with defining plaques that are not vulnerable and likely to do relatively well. This could be an exercise in identifying stable rather than vulnerable lesions or stable rather than vulnerable patients. Although the clinical relevance of the studies presented by Tarkin et al. may require years to mature, these studies contribute immensely to understanding and unraveling the pathogenesis of the disease process. After all, what could be more informative than the subcellular information obtained from a living organism vis-à-vis bench studies? The strength of the molecular imaging studies can hardly be underestimated.
_______
_______
Noninvasive assessment of endothelial function:
Flow-Mediated Dilation and Cardiovascular Event Prediction, a 2011 study:
Endothelial dysfunction is an early atherosclerotic event that precedes clinical symptoms and may also render established plaque vulnerable to rupture. Noninvasive assessment of endothelial function is commonly undertaken using the flow-mediated dilation (FMD) technique. Some studies indicate that FMD possesses independent prognostic value to predict future cardiovascular events that may exceed that associated with traditional risk factor assessment. It has been assumed that this association is related to the proposal that FMD provides an index of endothelium-derived nitric oxide (NO) function. Interestingly, placement of the occlusion cuff during the FMD procedure alters the shear stress stimulus and NO dependency of the resulting dilation: cuff placement distal to the imaged artery leads to a largely NO-mediated response, whereas proximal cuff placement leads to dilation which is less NO dependent. Authors used this physiological observation and the knowledge that prognostic studies have used both approaches to examine whether the prognostic capacity of FMD is related to its role as a putative index of NO function. In a meta-analysis of 14 studies (>8300 subjects), Authors found that FMD derived using a proximal cuff was at least as predictive as that derived using distal cuff placement, despite the latter being more NO dependent. This suggests that, whilst FMD is strongly predictive of future cardiovascular events, this may not solely be related to its assumed NO dependency. Although this finding should be confirmed with more and larger studies, Authors suggest that any direct measure of vascular (endothelial) function may provide independent prognostic information in humans.
______
______
Prevention and treatment of vulnerable plaque:
_
Prevention of vulnerable plaque:
Patients can lower their risk for vulnerable plaque rupture in the same ways that they can cut their heart attack risk: Optimize lipoprotein patterns, keep blood glucose levels low normal, stay slender, eat a proper diet, quit smoking, and maintain a regular exercise program.
_
Figure below shows algorithm for prevention of vulnerable plaque.
______
Diet and vulnerable plaque:
Direct association between diet and the stability of human atherosclerotic plaque: a 2015 study:
Coronary heart disease (CHD) mortality differs markedly across Europe and is generally lower in the southern than in the northern and eastern parts of the continent. Although the underlying causes for this difference remain to be clarified, there is emerging evidence that the Mediterranean diet contributes to the lower CHD mortality in southern Europe. Most acute coronary events are caused by thrombotic occlusion on top of a ruptured atherosclerotic plaque. The risk for plaque rupture is dependent on the structure of the plaque and rupture-prone or vulnerable plaques are characterized by enhanced inflammation, extensive lipid accumulation, large necrotic core, as well as loss of fibrous tissue and of the connective tissue-producing smooth muscle cells. To what extent dietary habits can influence CHD risk by direct effects on atherosclerotic plaque structure is not known. Using isotope ratio mass spectrometry (IRMS) it is possible to estimate the dietary origin of molecular components incorporated into biological tissues by analysing the composition of certain isotopes. The relative abundance of stable isotopes of carbon (expressed as 13C) and nitrogen (15N) can be used to differentiate between different types of terrestrial and marine-derived food.
Mediterranean diet has been suggested to explain why coronary heart disease mortality is lower in southern than northern Europe. Dietary habits can be revealed by isotope ratio mass spectrometry (IRMS) measurement of carbon (13C) and nitrogen (15N) in biological tissues. To study if diet is associated with human plaque stability, atherosclerotic plaques from carotid endarterectomy on 56 patients (21 Portuguese and 35 Swedish) were analysed by IRMS and histology. Plaque components affecting rupture risk were measured. Swedish plaques had more apoptosis, lipids and larger cores, as well as fewer proliferating cells and SMC than the Portuguese, conferring the Swedish a more rupture-prone phenotype. Portuguese plaques contained higher 13C and 15N than the Swedish, indicating that Portuguese plaques were more often derived from marine food. Plaque 13C correlated with SMC and proliferating cells, and inversely with lipids, core size, apoptosis. Plaque 15N correlated with SMC and inversely with lipids, core size and apoptosis. This is the first observational study showing that diet is reflected in plaque components associated with its vulnerability. The Portuguese plaques composition is consistent with an increased marine food intake and those plaques are more stable than those from Swedish patients. Marine-derived food is associated with plaque stability.
The Portuguese, on average, consume more fish products (13.4%) than Swedes (7.8%). The Portuguese diet includes more fish (leading to higher 13C and 15N values) and maize (C4 group, meaning higher 13C values), than the Swedish diet, which in general contains a significant amount of potatoes (C3 group, leading to lower values of both 13C and 15N), as well as other C3 foodstuffs (such as wheat, oats, rice, etc.). The 13C and 15N values now measured in the plaque samples further support this epidemiologic data.
In author’s’ sample collection, carotid plaques from Portuguese patients have a more stable histological phenotype than the Swedish, showing less apoptosis, lower lipid content, smaller cores, as well as more proliferating cells and smooth muscle cells, that stabilize the fibrous cap, avoiding rupture and the development of symptoms.
Taken together, these data indicate that the atherosclerotic plaque composition is consistent with the intake of certain dietary atoms. The composition of the Portuguese atherosclerotic plaques is associated with the increased intake of seafood and those plaques are more stable than those from Swedish patients. This study is pioneer in showing that marine-derived food is associated with plaque stability. Despite the need for further studies, the ultimate clinical implication of this knowledge is to encourage a simple and cheap strategy, as marine dietary intake in prevention of atherosclerosis, to stabilize rupture-prone plaques that ultimately lead to myocardial infarction and stroke.
______
______
Local Therapy for Vulnerable Plaque:
Current management guidelines focus on identifying and revascularizing obstructive coronary stenoses that cause a hemodynamically significant reduction in coronary blood flow. Despite extensive validation of this flow-limiting–guided revascularization strategy in large prospective randomized trials, such as the FAME and DEFER trials (Fractional Flow Reserve Versus Angiography for Multivessel Evaluation; Deferral of Percutaneous Coronary Intervention), the event rate after complete revascularization remains relatively high with MACE occurring in ≈30% of patients after 5 years (FAME trial). Serial angiography studies have shown that, although plaques leading to ACS are frequently flow limiting, coronary lesions with initially mild stenosis often rapidly progress in the weeks to months before an ACS.
_
The sine qua non of local therapy for vulnerable plaque is the existence and prospective detection of local areas of the vessel that are at increased risk of causing a coronary event. If the entire artery is found to be at high risk or if vulnerability rapidly emerges and resolves, there may be no role for local therapy. However, local diagnosis might still be of value as a guide to the intensity of systemic therapy, as is the case for cancer in which results of a local biopsy often guide systemic therapy.
In the context of uncertainty about the existence, detection, and timing of plaque vulnerability, it is not possible to recommend routine use of the promising local therapy options discussed below. Such therapy can be recommended only with the use of a vulnerability detector and the availability of data from a randomized trial demonstrating that the local therapy under consideration provides benefits greater than the complications associated with the intervention. Evaluation of the promising local treatments for vulnerable plaque (stents, photodynamic therapy, and other options) is therefore being hindered by the absence of validated diagnostic devices. For the vulnerable plaque hypothesis, detection of risk appears to be a more formidable obstacle than availability of possible treatments.
_
Treatment of Coronary Artery Risk Based on Location of Culprit Lesions:
Although technologies to identify risk caused by an individual plaque are not yet validated, Wang et al have identified regions of risk in arteries based on location of culprit lesions. It was found that most of the lesions causing the acute coronary syndromes occur within the proximal 30 mm of the major coronary arteries. These are also areas of decreased shear stress and increased numbers of TCFAs and ruptured plaques. Because the risk is associated with a relatively limited length of artery, it is possible that regional therapy (possibly with stents) might be useful for preventing subsequent events in patients undergoing PCI. Wang and colleagues have suggested that a randomized trial of such an approach might be conducted in high-risk patients. The trial might also use a vulnerability detector to identify patients with vulnerable arteries who would be the most likely to derive benefit.
_
Stents for the Treatment of Vulnerable Coronary Plaques and Arteries:
Drug-eluting stents have been evaluated in an atherosclerotic rabbit model as a possible treatment for vulnerable plaques. Placement of a stent over lesions in the aorta reduced lipid core size and induced formation of an additional fibrous cap over the lipid pool. Although these results are encouraging, the benefits of stenting vulnerable plaques in patients must be shown in a randomized trial to exceed the complications, which include periprocedural infarction, restenosis, and stent thrombosis.
_
Stenting for Intermediate Coronary Artery Stenoses That Are Not Flow Limiting:
There is generally little difficulty with the decision to stent a culprit lesion. It is causing or has caused ischemia, and the need for the stent is relatively straightforward and indicated by guidelines. Unfortunately, many patients also have a second or third stenosis that is not producing ischemia but causes concern because it is suspected to be vulnerable. The concern is heightened if the lesion causes a 50% to 60% diameter stenosis, has a complex angiographic appearance, and is in a precarious location such as the proximal left anterior descending coronary artery. In the absence of information about the likelihood that the plaque of concern might rupture, current guidelines call for the use of medical therapy. However, if the lesion were known to be vulnerable to rupture, stenting might be the better choice, depending on the balance of risks and benefits of stenting noted above.
The use of stents or angioplasty for lesions causing an intermediate stenosis was examined in the Defer Trial. Patients with intermediate stenoses not causing a significant reduction in fractional flow reserve were randomly assigned to intervention (angioplasty or stenting) or medical therapy. In a 5-year follow-up, there was no difference in the low rate of cardiac death and MI between patients assigned to medical therapy and those treated with the intervention. It was concluded that the low risk of MI and cardiac death in this population (<1% per year) did not justify intervention for intermediate stenoses in this group of patients. However, the study excluded patients with more active disease who had sustained an MI or unstable angina and did not use a vulnerability detector. The possibility that stenting of intermediate lesions suspected to be vulnerable will be of value in acute coronary syndrome patients has led to a proposal to conduct the Prevail Trial, a randomized trial of the strategy. Improvements in stent technology are likely to reduce the risks associated with stenting and thereby shift the risk-to-benefit ratio toward the stenting of intermediate stenosis.
_
Role of Vulnerability Detection in the Selection of Stenting Versus CABG as Optimal Therapy:
In several commonly encountered clinical situations, knowledge of the vulnerability of the artery in question might be useful as a guide to treatment with stents or referral for surgery. For diabetics with stenosis in 3 vessels, the current standard of care is CABG because of documented reductions in cardiac events with surgery that are not achieved by multivessel stenting. The bypass graft, which is attached distal to much of the epicardial coronary artery, presumably bypasses nonstenotic vulnerable plaques that would otherwise be responsible for subsequent events. Stenting of flow-limiting lesions, which is a focal treatment, would not provide such a benefit. However, if it were possible to determine whether the arteries contained vulnerable nonstenotic plaques, the triage of such patients to stenting or CABG could be improved. Patients with stenoses and no evidence of nonstenotic vulnerable plaques could be treated with stenting, and those with extensive signs of vulnerability could be sent for CABG. Multiple trials are in progress to compare the value of multivessel stenting with surgical treatment of patients with stenosis in 3 coronary vessels. Such trials would be aided by the detection of vulnerability and assessment of the degree of stenosis. Similar considerations apply to therapy for patients with isolated left main coronary artery stenosis. If a localized left main stenosis is present and the remainder of the arteries show no evidence of significant stenosis or vulnerability, stenting might be the preferred therapy. Alternatively, if there were signs of diffuse vulnerability throughout the artery, CABG might be preferred.
_
PTCA for the Treatment of Vulnerable Coronary Plaques:
In 1995, Meier proposed that it might be of value to intentionally rupture presumably vulnerable intermediate stenoses with balloon angioplasty at the time of catheterization. Because the rupture would occur under controlled conditions and in the presence of intensive antithrombotic therapy, the procedure would not cause an acute coronary event. After the catheterization, the ruptured lipid-rich plaque would be expected to become a fibrotic plaque, and the danger of a coronary event would be eliminated. The procedure was called plaque sealing. The concept stimulated extensive debate but did not become an accepted practice. Plaque sealing, however, could be reconsidered in light of new technical developments. The availability of stents provides an excellent treatment for the problems of subacute occlusion that limited the value of PTCA for preventive purposes. In addition, it is possible that a vulnerability detector might be able to identify the lesions most in need of therapy. The use of PTCA to intentionally rupture nonstenotic vulnerable plaques would not carry the long-term risks of stenting. Plaque sealing and all local therapies under discussion require evaluation in randomized trials.
_
The PRAMI study (Preventive Angioplasty in Myocardial Infarction Trial) reported a reduced risk of adverse cardiovascular events after revascularization of residual CAD in STEMI patients after primary PCI of infarct artery. Given that ≈35% of intermediate lesions are hemodynamically significant, sealing of nonobstructive vulnerable plaques is a potential explanation of improved outcome in the PRAMI study. However, no definite evidence exists that this is indeed the mechanism responsible for the improved outcome. The first-in-human trial of attempted sealing of nonobstructive high-risk plaques was the SECRITT trial (vShield Evaluated at Cardiac Hospital in Rotterdam for Investigation and Treatment of TCFA). In this trial, a nitinol self-expanding stent was implanted in a nonobstructive VH-TCFA with thin-cap at OCT in 13 patients and revealed recovery of neocap thickness without complications at 6-month follow-up. Instead of placing a traditional bare-metal stent, the use of a bioabsorbable vascular scaffold (BVS) may avoid long-term detrimental effects such as late stent thrombosis, by dissolving ultimately and facilitating restoration of normal coronary physiology. Implantation of an everolimus-eluting BVS in a porcine coronary artery model showed complete dissolution of the struts after 3 years by both OCT and histology and showed a fibromuscular neointima covering the plaque. In an in-human study, Brugaletta et al revealed formation of a thick neointima in 58 patients who received an ABSORB BVS, with a mean neointima thickness of 210 µm after 6 months and 220 µm after 12 months. A recent post hoc analysis, using data from the ABSORB cohort B trial, the SECRITT trial and the Svelte first-in-man trial evaluated changes in plaque morphology after BVS and bare-metal stent implantation. Serial OCT showed development of a neointima over TCFA, both after BVS and bare-metal stent implantation, thus sealing the plaque and transforming the TCFA to a thick-cap fibroatheroma. Furthermore, after BVS implantation the neointima development continued after short-term follow-up without compromising luminal dimensions, whereas neointima formation after bare-metal stent placement caused reduction in luminal dimensions and ceased after 6 months. Although evidence is limited, these findings suggest the feasibility of BVS to seal nonobstructive vulnerable plaques. Currently, 3 randomized trials that address this issue are ongoing as seen in the figure below:
_
Ongoing Clinical Trial on the Preventive Local Treatment of Vulnerable Plaques
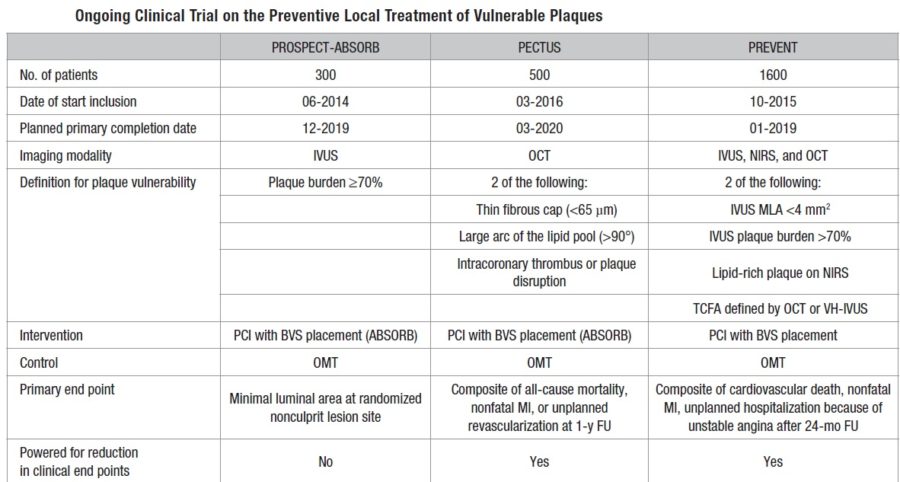
BVS indicates bioabsorbable vascular scaffold; FU, follow-up; IVUS, intravascular ultrasound; IVUS-NIRS, intravascular ultrasound-near-infrared spectroscopy; MI, myocardial infarction; NIRS, near-infrared spectroscopy; OCT, optical coherence tomography; OMT, optimal medical therapy; PCI, percutaneous coronary intervention; PECTUS, Pre-Emptive, OCT Guided Angioplasty of Vulnerable, Intermediate Coronary Lesions: A Randomized Trial; PREVENT, The Preventive Implantation of Bioresorbable Vascular Scaffold on Functionally Insignificant Stenosis With Vulnerable Plaque Characteristics; PROSPECT-ABSORB, A Multicentre Prospective Natural History Study Using Multimodality Imaging in Patients With Acute Coronary Syndromes Combined With a Randomized, Controlled, Intervention Study; TCFA, thin-cap fibroatheroma; and VH-IVUS, virtual histology intravascular ultrasound.
Recent meta-analysis of the 4 randomized ABSORB trials demonstrates that compared with metallic everolimus-eluting stents, BVS have higher rates of target lesion failure and device thrombosis cumulatively to 3 years and between 1 and 3 years.
______
Should we treat the vulnerable plaque (VP) with angioplasty?
About 90% of clinically relevant MI are caused by lesions in the proximal third of the major coronary arteries. An immediate decision is warranted whether to proceed with a preventive angioplasty to transform this vulnerable plaque mechanically into a scar tissue by intentionally cracking the fibrous cap under controlled condition to induce a scar which is immune to future fissures or rupture (plaque sealing). Thus once the acute effect with its inherent risk of acute occlusion has passed, the subsequent risk of plaque rupture is markedly reduced compared with that of an untreated plaque. There is little published evidence for the plaque sealing concept. Mercado et al have investigated the clinical and angiographic outcome of patients with mild coronary lesions treated with balloon angioplasty and stenting in comparison with moderate to severe stenosis. The one-year event rate (mortality, rate of non-fatal MI, and repeat revascularization) was same for significant and non-significant stenoses. The potential risk of performing angioplasty and for causing significant restenosis is such non-significant lesions should be taken into account. The advent of the drug eluting stents offers advantage in favour of mechanical sealing by reduction of restenosis rate.
Many thoughtful investigators have shown that plaque instability is a distinctly multifocal phenomenon. We are not likely to find “the” vulnerable plaque, because, in fact, a multitude of such high-risk lesions are likely present. Available data suggest that there may be dozens of lesions present, distributed throughout the coronary circulation. So, if we could find the vulnerable plaques, do we really think we could stent them all? If a handful of particularly high-risk lesions were detected and “sealed,” will the immediate risks and late adverse consequence of coronary intervention (late stent thrombosis) actually exceed any prospective benefits? The COURAGE (Clinical Outcomes Using Revascularization and Aggressive Drug Evaluation) trial suggests that the risks of such a strategy may exceed any benefits.
Use of stents to treat VP — that is causing some controversy among interventional cardiologists.
Dr. Poon said stents are sometimes used to stabilize the plaque by pushing it deeper into the wall. But he admitted, “You basically are sweeping it under the rug.” “I think it’s a misuse of stenting because we are not good enough yet in picking out plaques that are at high risk of rupturing,” Dr. Shapiro said. “Even if it’s a drug-eluting stent, the patient is at risk of developing a thrombus. And that’s too big a jump. There needs to be more data before that step is taken.” Dr. Sharma says a stent can lower the risk of a VP rupturing. Yet he added, “You really cannot treat VP using a stent. You need aggressive medical treatment.” Dr. Rutherford weighs in by saying that the major concern about using a stent is that you can accidentally rupture the plaque — thus creating the problem you wanted to avoid. But he also holds out hope for the future.
__
In a nutshell:
Imaging ‘vulnerable’ plaques is a fascinating research tool providing useful insights into the pathophysiology of ACS. At present, it has little clinical use. Local therapy, in the absence of documented ischaemia, is unproven.
__
Photodynamic Therapy for Vulnerable Coronary Arteries and Plaques:
Photodynamic therapy has been proposed as a method to stabilize a specific plaque or a region of an artery by selective ablation of macrophages or other targeted cells. The photosensitizing agent motexafin lutetium was tested in atherosclerotic rabbits. After photoactivation via an intra-aortic catheter, there was a marked reduction in the number of macrophages and a small decrease in atheroma burden without damage to normal tissue. The same agent has been administered to patients undergoing coronary stenting in a safety study and found to be well tolerated. Using a different photodynamic agent, Waksman et al demonstrated that photodynamic therapy can reduce neointimal growth without suppressing reendothelialization of a stent in a porcine model. These early results suggest that photodynamic therapy might eventually have a role in the local or regional treatment of vulnerability.
_
Cooling, Heating, and Sonotherapy:
Cryoenergy has been shown to reduce restenosis and increase the density of type III collagen in balloon-injured porcine arteries. It may be useful to stabilize vulnerable plaques because it can induce local apoptosis without causing excessive neointimal proliferation. Cryoenergy is currently being studied for the treatment of peripheral arterial disease. Although heating and sonotherapy have also been considered as possible methods to treat vulnerable plaque, little supporting evidence is available.
______
______
Systemic Therapy for Vulnerable Plaque:
Ambrose and D’Agate have proposed a useful classification to assess the likelihood that a given systemic therapy is plaque stabilizing as seen in the table below. Therapies are grouped by the strength of the evidence that they prevent coronary events and the plausibility of a mechanism by which they might do so by stabilizing vulnerable plaques.
| Classification of Systemic Therapy for Vulnerable Plaque | |
| Therapies | Drug |
| Group 1: therapies with biological plausibility and positive clinical evidence | Statins, ACE inhibitors, β-blockers, aspirin |
| Group 2: therapies with biological plausibility but negative clinical evidence | Antioxidants, folic acid, antibiotics |
| Group 3: therapies with biological plausibility but conflicting or inconclusive clinical evidence | Angiotensin-receptor blockers, omega-3 fatty acids, other antihypertensive agents, cyclooxygenase-2 inhibitors, influenza vaccine, clopidogrel, metalloproteinase inhibitors |
| Group 4: therapies with biological plausibility but no clinical data available | Peroxisome proliferator–activated receptor antagonists, cholesterol ester transfer protein inhibitors |
Although systemic therapy is clearly the preferred solution, current systemic therapy does not provide adequate protection, particularly for the very-high-risk group of patients whose disease activity has led to the need for a PCI.
__
While prospectively detecting and treating vulnerable plaque remains a challenge, some medications, current and emerging, can help stabilize plaque for secondary prevention, according to a European Society of Cardiology (ESC) position paper in 2011. The ESC Working Group of Atherosclerosis and Vascular Biology noted in the paper that strong clinical evidence has shown the ability of statins to stabilize plaque, along with positive results for aspirin and other antiplatelet agents, beta-blockers, and renin-angiotensin-aldosterone system inhibitors. Researchers also noted the existence of some evidence showing plaque stabilization abilities for peroxisome proliferator activated receptor (PPAR) agonists, niacin, omega-3 fatty acids, and some HDL-raising therapies.
_
Although local treatment of an individual vulnerable plaque is appealing, it fails to address the fact that atherosclerosis is a systemic disease. Various systemic therapies have been proposed to reduce plaque vulnerability as seen in the figure below:
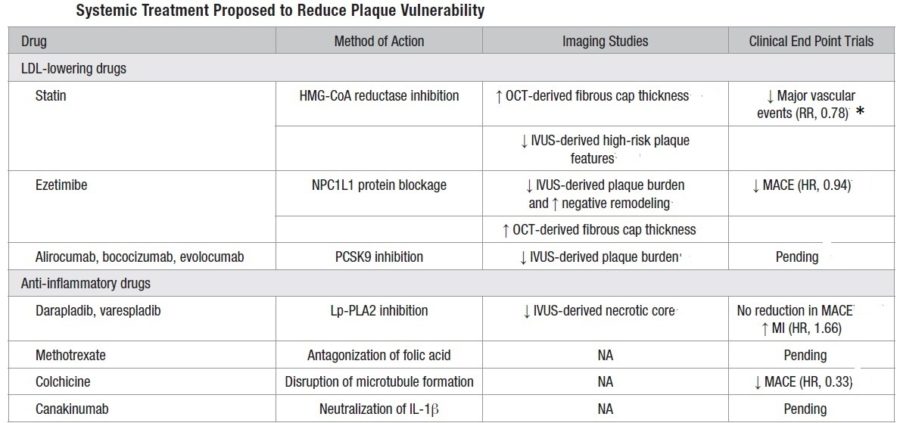
HMG-CoA indicates 3-hydroxy-3-methylglutaryl-coenzyme A; HR, hazard ratio; IL-1β, interleukine-1β; IVUS, intravascular ultrasound; LDL, low-density lipoprotein; Lp-PLA2, lipoprotein-associated phospholipase A2; MACE, major adverse cardiac events; MI, myocardial infarction; NA, not available; NPC1L1, Niemann-Pick C1-like 1; OCT, optical coherence tomography; PCSK9, proprotein convertase subtilisin/kexin type 9; and RR, relative risk.
*Meta-analysis of 26 randomized trials showing reduction in major vascular events.
_
Drugs that may Enhance Plaque Stability:
A number of drugs that are beneficial for patients with coronary disease may act in part by improving the stability of plaques that are vulnerable for future rupture.
-Beta blockers lower circumferential wall stress via reductions in blood pressure, heart rate, left ventricular mass, and catecholamine surges that occur with stress and exertion.
-Angiotensin converting enzyme (ACE) inhibitors lower the blood pressure and may have plaque-stabilizing effects by reducing the synthesis of angiotensin II.
-Lipid-lowering therapy, particularly with statins, can stabilize vulnerable plaques or those that have already ruptured by improving endothelial function and reducing thrombogenicity, platelet aggregation, and possibly inflammation.
– Antithrombotic therapy, including antiplatelet agents and warfarin do not stabilize the vulnerable plaque, but they do limit thrombosis, which is an important consequence of plaque rupture.
___
Potential benefits of eicosapentaenoic acid on atherosclerotic plaques, a 2017:
Residual cardiovascular (CV) risk remains in some patients despite optimized statin therapy and may necessitate add-on therapy to reduce this risk. Eicosapentaenoic acid (EPA), an omega-3 polyunsaturated fatty acid, lowers plasma triglyceride levels without raising low-density lipoprotein cholesterol levels and has potential beneficial effects on atherosclerotic plaques. Animal studies have shown that EPA reduces levels of pro-inflammatory cytokines and chemokines. In clinical trials utilizing a wide spectrum of plaque imaging modalities, EPA has shown beneficial effects on plaque characteristics. Studies of patients with coronary artery disease receiving statin therapy suggest that EPA may decrease plaque vulnerability and prevent plaque progression. EPA also decreased pentraxin-3 and macrophage accumulation. A large, randomized, Japanese study reported that EPA plus a statin resulted in a 19% relative reduction in major coronary events at 5 years versus a statin alone in patients with hypercholesterolemia (P = 0.011). Icosapent ethyl, a high-purity prescription form of EPA ethyl ester, has been shown to reduce triglyceride levels and markers of atherosclerotic inflammation. Results of an ongoing CV outcomes study will further define the potential clinical benefits of icosapent ethyl in reducing CV risk in high-risk patients receiving statin therapy.
___
Anti-Inflammatory Drugs:
Inflammation plays a key role in plaque formation and vulnerability. Several anti-inflammatory therapies have been proposed to reduce atherosclerotic burden and stabilize coronary plaques. Lipoprotein-associated phospholipase A2 is an enzyme produced and secreted by inflammatory cells centrally involved in atherosclerosis and is highly expressed in the necrotic core of atherosclerotic plaques. Lipoprotein-associated phospholipase A2 inhibition has been proposed as a therapeutic agent targeting the vulnerable plaque. Although a randomized placebo-controlled trial reported a reduction in IVUS-derived necrotic core development, large randomized trials on the benefit of lipoprotein-associated phospholipase A2 inhibitors on clinical end points have to date been negative. Methotrexate is a folic acid antagonist with anti-inflammatory actions and is used for the treatment of several autoimmune disorders. A large meta-analysis of observational studies in patients receiving low-dose methotrexate for psoriasis and rheumatoid arthritis showed a reduction in cardiovascular events. The CIRT (Cardiovascular Inflammation Reduction Trial) was designed to investigate the ability of low-dose methotrexate to prevent future coronary events in patients after MI. The anti-inflammatory activity of colchicine is used in the treatment of inflammatory conditions such as gout, pericarditis, and familial Mediterranean fever. Retrospective studies have shown indirect support for the use of colchicine in atherosclerosis by reporting lower prevalence of MI in colchicine-treated patients having gout and familial Mediterranean fever. This was confirmed by the LoDoCo trial (Low-Dose Colchicine), a randomized placebo-controlled trial with 532 patients with stable CAD and median follow-up of 3 years, showing improved MACE-free survival in patients treated with colchicine. The LoDoCo-2 trial will investigate the effects of colchicine in 3000 patients over 3 years and is scheduled to be completed in 2019. Doxycycline at a common antimicrobial dose stabilizes atherosclerotic lesions via inhibiting matrix metalloproteinases and attenuating inflammation in a rabbit model of vulnerable plaque. These effects were similar to a large dose of simvastatin and independent of serum lipid levels.
Canakinumab is a monoclonal antibody that selectively neutralizes interleukin-1β, a proinflammatory cytokine that plays multiple roles in atherosclerosis and rupture prone plaques. A recent phase IIb trial with 556 patients has shown that subcutaneous injection of canakinumab is well tolerated and reduces different biomarkers of inflammation, that is, C-reactive protein, interleukin-6 and fibrinogen. The CANTOS trial (Canakinumab Anti-Inflammatory Thrombosis Outcomes Study) has finalized enrollment of 101,20 patients and is awaiting follow-up to report on the possible reduction of recurrent MACE by canakinumab in patients after MI.
____
Therapies to Increase HDL Cholesterol:
It is likely that increases in HDL would increase plaque stability because such changes accelerate reverse cholesterol transport, lowers oxidation level, and decreases inflammation. Clinical studies have documented the prevention of coronary events with HDL raising by niacin and gemfibrozil therapy. With interest in HDL-raising therapies, niacin has been recently re-investigated. Two trials in statin-treated patients with low HDL have shown that modified-release nicotinic acid significantly reduced carotid atherosclerosis, and that the use of slow-release niacin significantly reduced carotid intima-media thickness in comparison with statin. However, AIM-HIGH and HPS-2-THRIVE trials failed to show any added benefits which raises doubts about the usefulness of this approach. Inhibitors of cholesteryl ester transport protein have emerged as promising agents to increase HDL levels. Torcetrapib and JTT-705 can produce elevations of ≥50% in HDL cholesterol. Clinical trials are in progress to determine whether such HDL elevations produce the expected clinical benefits. The atheroprotective effects of apoA-1 Milano, a synthetic form of HDL, have received considerable attention. The agent was infused weekly for 5 weeks into patients recovering from an acute coronary syndrome. IVUS measurements demonstrated a small decrease in atheroma volume, supporting the transition to studies with clinical end points. Additional approaches in this active field include the use of small molecules to increase HDL levels and a variety of nuclear hormone receptor agonists to increase ABC gene transcription and HDL levels and/or function. There are also efforts to evaluate systemic administration of unilamellar phospholipid vesicles and gene therapy involving HDL-related proteins.
______
Antiplatelet and antihypertensive therapies:
Aspirin is effective for CVD secondary prevention, and a major reduction in CVD events was found in the CURE trial, where clopidogrel was added to aspirin in ACS patients. However, these agents mostly reduce complications of plaque rupture and may not be plaque-stabilizing agents per se. Whether new platelet inhibitors ticagrelor and prasugrel and platelet thrombin receptor inhibitor vorapaxar have plaque-stabilizing properties remains unknown, although recent meta-analyses have suggested the same benefits in patients with recent ACS.
Four recent IVUS trials have shown that β-blockers slow the progression of CVD. Endothelial function can be improved by renin–angiotensin inhibitors, and HOPE and ONTARGET trials have shown a larger reduction in CVD events that could be predicted from the reduction in blood pressure, supporting plaque-stabilizing effects.
_____
_____
Statins:
In recent years primary and secondary prevention trials have unequivocally proved that reduction in cholesterol using 3-hydroxy-3-methylglutaryl coenzyme A (HMGCoA) reductase inhibitors (the statins) reduces subsequent clinical events on average by 30%–40%. HMGCoA reductase catalyses the rate limiting enzyme in cholesterol biosynthesis, and drug treatment reduces serum low density lipoprotein (LDL) cholesterol in patients. Although many of the beneficial effects of statins may be via reduction in cholesterol, some effects of these drugs appear to be independent of LDL cholesterol lowering. Thus, benefit from statins occurs in low risk patients with relatively “normal” cholesterol levels. In addition, administration of pravastatin to lipid fed monkeys at doses that did not reduce cholesterol levels still reduced macrophage content of plaques. Statins also have potent effects on inflammation, both locally within the plaque and systemically. Statins reduce the macrophage content of atherosclerotic plaques and the activity of the residual macrophage populations, and some statins also reduce serum C reactive protein and interleukin-6 in patients, independent of lipid lowering. In addition, statins reduce activity of natural killer cells and inhibit antibody production. Statins also inhibit MMP-9 expression of both mouse and human macrophages, and have variable effects on matrix proteins and collagen. Oxidised LDL induced growth of macrophages and foam cell formation is also inhibited by statins.
_
The mortality benefit of LDL-lowering therapy by use of statins is well established, and its use in patients with CAD is advocated by current guidelines. In addition to their lipid-lowering properties, statins are reported to possess pleotropic effects, that is, enhancement of endothelial function and decrease of oxidative stress and inflammation. The effects of statins on coronary plaque progression in vivo have been investigated by various imaging modalities. Serial CCTA studies have shown that initiation of statin therapy reduces progression of noncalcified plaque volume and even progression in LAP volume. Furthermore, a recent trial randomized patients between high-intensity and standard statin therapy showed increase in dense calcium volume, implicating possible plaque stabilization.
Several serial IVUS studies have shown that high-intensity statin therapy induces regression in plaque burden. This was recently confirmed by a post hoc analysis including data from 3495 patients. In addition to reduction of plaque burden, the authors reported that statin therapy induces plaque calcification, which is associated with plaque stabilization. Kataoka et al published pooled data from 8 clinical trials on the beneficial effect of statins in IVUS-derived high-risk plaques (large atheroma volume, positive remodeling, and spotty calcification). The authors reported that atheroma regression was highest in patients with high-risk plaques, suggesting a plaque-stabilizing effect of statin therapy. This is supported by findings from OCT studies. Kataoka et al reported that high-intensity statin therapy in patients with stable CAD is associated with less vulnerable plaque features on OCT, that is, thicker fibrous cap and smaller lipid arc. Furthermore, the EASY-FIT study (Effect of Atorvastatin Therapy on Fibrous Cap Thickness in Coronary Atherosclerotic Plaque as Assessed by Optical Coherence Tomography) showed that in patients with unstable angina high-intensity statin therapy provided greater increase in fibrous cap thickness than low-intensity statin therapy. Because this increase in cap thickness was associated with decrease in inflammatory biomarkers, it is believed that in addition to the LDL-lowering action, anti-inflammatory effects of statin therapy may influence plaque vulnerability. Nissen et al studied the effect of intensive lipid lowering with rosuvastatin on coronary atheroma in 507 patients undergoing IVUS examination at baseline and after 2 years of follow-up. The marked reduction in LDL was associated with an 11.1% decrease in coronary plaque volume as measured by IVUS. Although the study was not randomized and HDL increases also occurred, it is quite likely that the intense LDL lowering produced this beneficial decrease in plaque volume and stabilized plaques. The German Atorvastatin Study demonstrated that hyperechogenicity of plaques significantly increased after 12 months compared with non-statin-based lipid lowering. In the ASTEROID and SATURN studies, aggressive lipid lowering regressed atheroma volume in IVUS. Data from several prospective IVUS trials confirmed significant atheroma regression after LDL reduction and the PROVE-IT trial showed lower CVD endpoints after 24 months under intense lipid lowering in ACS patients. In addition, the JUPITER study in healthy subjects with LDL <3.4 mmol/L and hs-C-reactive protein above 2 mg/L showed that 20 mg of rosuvastatin significantly reduced major CVD events in this low-risk group.
_____
Effect of statin therapy on the progression of coronary atherosclerosis, a 2012 meta-analysis:
An increasing number of authors employing intravascular ultrasound (IVUS) and virtual histology (VH-IVUS) have investigated the effect of statin use on plaque volume (PV) and plaque composition. However, inconsistent results have been reported. Therefore, authors conducted a meta-analysis to determine the appropriate regimen of statins to effectively stabilize vulnerable coronary plaques. The meta-analysis demonstrated that statin therapy (especially that involving a high dose and long duration and achieving <100 mg/dL LDL-C levels) can significantly decrease PV in patients with stable angina or ACS. These data suggested that statins can be used to reduce the atheroma burden for secondary prevention by appropriately selecting the statin regimen. No significant change in plaque composition was seen after statin therapy.
_
Study selection is summarized in figure below:
_
This present study demonstrated that statin therapy can significantly decrease the size of atherosclerotic plaques, whereas there were no significant changes in plaque composition. These findings are similar to the findings in some in-vivo human studies. The findings are supported by animal studies, which have demonstrated a reduction in plaque size and macrophage content as well as an increase in the amount of interstitial collagen in atherosclerotic lesions after lipid-lowering therapy.
Plaque composition is now seen as being much more important than plaque size and the severity of stenoses. Plaques containing a soft lipid-rich core are particularly dangerous because such plaques are unstable and vulnerable to rupture, whereby highly thrombogenic plaques are exposed to blood flow. Plaques that are prone to rupture have a large lipid core, occupy >40% of the total PV, and have a thin fibrous cap (diameter, <65 μm). Reduction in the size of the necrotic core might be one of several requirements for plaque stabilization. After statin therapy, the necrotic core tended to decrease in size, but this did not reach statistical significance. Some studies using optical coherence tomography (OCT) showed that the incidence of plaque rupture was significantly decreased and that the thickness of the fibrous cap tended to increase with statin therapy.
Some researchers have paid close attention to the pleiotropic effects of statins that are independent of lipid lowering effects. These include immune modulation, inhibition of the proliferation and migration of smooth muscle cells, and inhibition of angiogenesis. The primary action of statins is a cholesterol-dependent effect: inhibition of the synthesis of L-mevalonate (a precursor of cholesterol). The secondary actions are pleiotropic: inhibition of the synthesis of isoprenoids such as farnesyl pyrophosphate and geranylgeranyl pyrophosphate (which are essential for membrane translocation and the biological activity of members of the GTPase family). Williams et al. documented that pravastatin-treated monkeys had atherosclerotic lesions with less neovascularization and less infiltration of macrophages independent of plasma concentrations of lipoproteins. Atorvastatin treatment has been shown to dose-dependently reduce the level of a serum marker of oxidative stress. Furthermore, those pleiotropic effects of statins are often larger with high doses of statins, and there may be individual differences among different statins. The present study has shown that regimens employing high doses of statin result in plaque regression in subjects with CHD. The dose-dependent pleiotropic effects of statins may be one of the explanations for authors’ findings. In the present study, greater benefit was noted among patients with ACS, which led to an improvement in outcome. The pleiotropic effects of statins (particularly their anti-inflammatory and antithrombotic properties) have been invoked to explain the greater benefits seen in ACS. However, it remains unclear which statin has the greatest and most rapid pleiotropic effects. Large, prospective, controlled studies are needed to determine the role of the pleiotropic effects of statins on atherosclerosis.
________
Impact of statin therapy on coronary plaque composition: a systematic review and meta-analysis of virtual histology intravascular ultrasound studies, 2015:
A new meta-analysis by Banach et al. reinvestigated the effect of statins on atherosclerosis progression and was recently published in BMC Medicine. The authors included nine studies with more than 830 individuals in whom virtual histology data was available. This study supported previous findings that higher statin doses result in significant plaque volume reduction, while lower statin doses does not. More interestingly, the pooled virtual histology data demonstrated that this small change in plaque volume is a poor summary of plaque composition change. On the one hand, the fibro-fatty and necrotic core volumes remained unchanged with statin use; and on the other hand, a significant fibrous plaque volume reduction accompanied a significant increase in the dense calcium volume. The data also suggests that there is a direct correlation between statin intensity used in each study and the effects on fibrous plaques and on dense calcium volumes, despite the limited power of subgroup analysis. Another very recent meta-analysis reached similar conclusions using different analytical techniques.
An interesting aspect of plaque composition derived from the study by Banach et al. is the role of coronary calcification in the atherosclerotic process and its implications on cardiovascular risk assessment. While some authors propose that calcium formation is part of the healing and stabilizing process of atherosclerosis, other in vitro studies indicate that calcium location may better explain the difference in plaque rupture risk. In fact, while clinical studies have firmly documented a direct association between the overall calcification in coronary arteries (measured as either the Agatston score or calcium volume) and cardiovascular events, other studies suggest that the pattern and distribution of calcium in coronary plaques may equally matter. A classical coronary computed tomography (CT) angiography study by Motoyama et al. indicated that small “spotty” calcifications are associated with future plaque ruptures, whereas a sub-analysis of the Multi-Ethnic Study of Atherosclerosis (MESA) study demonstrated that calcium density is inversely associated with events risk. These results, grounded by cellular and molecular data on the mechanisms regulating the pattern of atherosclerotic calcification, suggest that smaller calcium deposition (pro-inflammatory-driven microcalcifications) are associated with plaque rupture and increased cardiovascular risk, while larger, denser calcium structures (anti-inflammatory-driven macrocalcifications) are associated with plaque stabilization and better outcomes, in agreement with the increased dense calcification documented with statin treatment.
Collectively, these results indicate that instead of regression, the use of statins can lead to plaque healing and stabilization. The “healed” plaque is only discretely smaller, although it has better structure and is less prone to rupture. Even so, the occurrence of cardiovascular events despite optimized treatment in a significant proportion of patients receiving statins, the so-called “residual risk”, suggests that the healing process is incomplete. In fact, it has been proposed that statins and other preventive measures alter the mechanisms leading to acute coronary syndromes. Instead of plaque rupture, acute coronary syndromes in those “healed” plaques are more likely to occur by erosion. This change is thought to be related to the improved plaque structure associated with the change in its components.
____
Changes in Plaque Composition and the Pleiotropic Effects of Statins, 2016 study:
Park et al. in the STABLE (Statin and Atheroma Vulnerability Evaluation) trial found that rosuvastatin reduced necrotic core and plaque volume and decreased thin-cap fibroatheroma rate. However, there were no significant differences between high (40 mg) – versus moderate (10 mg) – intensity rosuvastatin, thus questioning that change in plaque composition is mediated by the low-density lipoprotein (LDL) cholesterol lowering effect of rosuvastatin.
Interestingly, a diminution of the risk gradient between cholesterol levels and coronary heart disease (CHD) events in the recent statin era has been found. Authors suggest both the change in plaque composition, irrespective of statin induced LDL lowering, and changes in the risk gradient between cholesterol levels and CHD may represent the proof of concept of the multiple biological activities of statin drugs, often referred to as “pleiotropic” effects.
It is well known that not all drugs that improve lipid profiles reduce patient risk. In fact, the trial evidence indicates that risk is not reduced by treatment to target, but by the use of statins. Indeed, in patients with familial hypercholesterolemia, lower statin doses than those currently advised have been shown to reduce the risk of CHD to a greater extent than anticipated, thus suggesting that even in familial hypercholesterolemia, the effects of statins may be mediated by their pleiotropic action.
Recently, in a pre-specified analysis of the IMPROVE-IT (Improved Reduction of Outcomes: Vytorin Efficacy International Trial), the relationship between achieved LDL and high-sensitivity C-reactive protein (hs-CRP) targets and outcomes for patients randomly assigned to simvastatin monotherapy and the combination of ezetimibe/simvastatin was analyzed. It was observed that significantly more patients met both of the pre-specified targets of LDL <70 mg/dl and hs-CRP <2 mg/l in the ezetimibe/simvastatin group in comparison with simvastatin alone, and that achievement of both LDL-C and hs-CRP targets was associated with significantly improved cardiovascular outcomes in comparison with those meeting neither target after multivariable adjustment. Even though the addition of ezetimibe increased the likelihood of targets attainment, the benefit in risk reduction for those meeting both targets was similar by treatment arm. These results clearly indicate that the risk reduction was essentially driven primarily by simvastatin action, the only drug present in both randomized groups.
Therefore, change in plaque composition irrespective of the LDL lowering action of rosuvastatin should be interpreted as the confirmation that statins in part exert their beneficial effects in reducing cardiovascular risk through their pleiotropic action, and that cardiovascular prevention should no more be considered simply a “cholesterol issue.”
_
Therapeutic effects of different Atorvastatin doses on vulnerable plaques in coronary arteries assessed by intracoronary optical coherence tomography, a 2018 study:
The aim of this study was to evaluate optical coherence tomography (OCT) as an assessment of the efficacy of atorvastatin treatment. Twenty-four acute coronary syndrome (ACS) patients were allocated to conventional-dose (20 mg atorvastatin, n = 12) and intensive-dose (40–80 mg atorvastatin, n = 12) groups and correlations between changes in the OCT measurements and blood routine indexes were analyzed 9 months post-percutaneous coronary intervention (PCI). Treatment with atorvastatin resulted in a significant increase in the target thin cap fibroatheroma (TCFA) fibrous cap thicknesses in both groups. The increase was bigger in the intensive-dose group than in the conventional-dose group (184.1 ± 57.4 μm vs. 125.1 ± 28.6, P = .005). The TCFA lipid core arc in both groups was significantly decreased compared with baseline (72.9 ± 29.3 vs. 127.6 ± 50.8, P < .01 and 74.6 ± 32.9 vs. 132.6 ± 51.3, P < .01, respectively). Correlation analyses showed an inverse relationship between low-density lipoprotein cholesterol (LDL-c) levels and the TCFA cap thickness, and a direct relationship between C-reactive protein (CRP) level and lipid core arc. Statins significantly increased the TCFA fibrous cap thickness and reduced the lipid core arc, and OCT measurements accurately reflected the levels of blood LDL-c and CRP.
MIRACLE and PROVE-IT studies have shown that early treatment of ACS patients with atorvastatin at an intensive dose could significantly decrease the incidence of cardiac events. The ESTABLISH study reported a successful reversal of atherosclerotic coronary plaques by treatment with atorvastatin at a moderate dose (20 mg). Furthermore, a reversal study demonstrated that atorvastatin at an intensive dose (80 mg) rather than a conventional dose (40 mg) was more effective in the prevention of atherosclerotic plaque progression. However, the latter 2 studies assessed the plaque volume and composition by virtual histology intravascular ultrasound (VH-IVUS), which failed to measure accurately the fibrous cap thickness and lipid core arc of the plaques owing to the relatively low resolution of this method. In this study, the effects of atorvastatin at conventional and intensive doses were evaluated by directly measuring the TCFA fibrous cap thickness and lipid core arc utilizing OCT, and the therapeutic strategies with statins for ACS patients were further assessed.
This study showed that treatment with atorvastatin both at conventional and intensive doses significantly increased the thickness of the fibrous cap and reduced the lipid core arc in vulnerable plaques. Moreover, the increase in the thickness of the fibrous cap was significantly greater in the intensive-dose group compared to the conventional-dose group. The thickness of the thinnest fibrous cap inversely correlated with the LDL level, and the degree of the lipid core arc directly correlated with the CRP level, suggesting a dual effect of atorvastatin. OCT is likely to be an excellent intracoronary detection method and can be used to evaluate the therapeutic effects of statins.
_
Association Between Baseline LDL-C Level and Total and Cardiovascular Mortality After LDL-C Lowering
A Systematic Review and Meta-analysis, 2018:
Question
Does the magnitude of reductions in total and cardiovascular mortality after low-density lipoprotein cholesterol (LDL-C) lowering depend on the baseline LDL-C level?
Findings
In this meta-analysis of 34 randomized clinical trials that included 270 288 participants, more intensive LDL-C–lowering therapy was associated with a progressive reduction in total mortality with higher baseline LDL-C levels (rate ratio, 0.91 for each 40-mg/dL increase in baseline level); however, this relationship was not present with baseline LDL-C levels less than 100 mg/dL. There was a similar relationship for cardiovascular mortality.
Meaning
The greatest benefit from LDL-C–lowering therapy may occur for patients with baseline LDL-C levels of 100 mg/dL or greater.
Conclusions and Relevance
In these meta-analyses and meta-regressions, more intensive compared with less intensive LDL-C lowering was associated with a greater reduction in risk of total and cardiovascular mortality in trials of patients with higher baseline LDL-C levels. This association was not present when baseline LDL-C level was less than 100 mg/dL, suggesting that the greatest benefit from LDL-C–lowering therapy may occur for patients with higher baseline LDL-C levels.
_
Statins for primary prevention of cardiovascular events and mortality in old and very old adults with and without type 2 diabetes: retrospective cohort study, 2018:
In participants older than 74 years without type 2 diabetes, statin treatment was not associated with a reduction in atherosclerotic CVD or in all-cause mortality, even when the incidence of atherosclerotic CVD was statistically significantly higher than the risk thresholds proposed for statin use. In the presence of diabetes, statin use was statistically significantly associated with reductions in the incidence of atherosclerotic CVD and in all cause mortality. This effect decreased after age 85 years and disappeared in nonagenarians.
_
Association of Statin Adherence with Mortality in Patients with Atherosclerotic Cardiovascular Disease, 2019 study:
Using a national sample of Veterans Affairs patients with ASCVD, authors found that a low adherence to statin therapy was associated with a greater risk of dying. Women, minorities, younger adults, and older adults were less likely to adhere to statins. Their findings underscore the importance of finding methods to improve adherence.
_
Q and A:
Q: Is the anti-inflammatory effect of statins due to lipid lowering or is it an independent effect?
A: The evidence suggests that lipid lowering is the dominant mechanism, because most of the changes in plaque volume occur around the time that lipid lowering has already occurred. However, some studies showed anti-inflammatory effects independent of lipid lowering. In the animal model there is some data to suggest that anti-inflammatory effects are dissociated from lipid-lowering effects.
Q: Do the findings from the carotid arteries apply to coronary arteries?
A: It is likely that what occurs in one area of the circulation applies to other areas of the circulation. However, it is difficult to be absolutely certain. Coronary plaques are more difficult to assess in this manner, so we are limited to using extra-coronary sites to gain some insights. The findings are consistent with other clinical observations.
Q: How low should LDL be lowered?
A: Findings from clinical trials support the idea that reducing LDL well below 100 mg/dL can have positive payoffs. Trials with acronyms such as PROVE-IT, REVERSAL, ASTEROID, and JUPITER have shown that using powerful statins such as atorvastatin or rosuvastatin to lower LDL to 70 mg/dL or below halted or even reversed the steady spread of atherosclerotic plaque and also reduced the rate of heart attack and stroke. It is still not clear how low LDL should be reduced, as it is only one of the risk factors. There are patients who develop MI and stroke who have perfectly normal LDL levels. Thus, there may be a threshold below which little additional benefit is seen. Whether that level is 100 or 70 is still subject to question. When baseline LDL level is less than 100 mg/dL, statins are not reducing mortality. This challenges pleiotropic actions of statins.
_______
Besides statins, other agents to reduce LDL levels:
The IMPROVE-IT trial (Improved Reduction of Outcomes: Vytorin Efficacy International Trial) with a total number of 18,144 patients showed a reduction in adverse cardiac events in patients on ezetimibe and simvastatin versus simvastatin only and paved the way for the introduction of ezetimibe into treatment guidelines. A randomized clinical trial with serial IVUS imaging showed that combination therapy of ezetimibe and statin was correlated with negative vessel remodeling and produced stronger coronary plaque regression than statin therapy alone. A small single-center randomized study showed that addition of ezetimibe to fluvastatin resulted in a larger increase in fibrous cap thickness measured by OCT.
Recently, a novel class of LDL-lowering drugs called proprotein convertase subtilisin–kexin type 9 inhibitors has emerged. Proprotein convertase subtilisin/kexin type 9 (PCSK9) is an enzyme encoded by the PCSK9 gene in humans on chromosome 1. PCSK9 is ubiquitously expressed in many tissues and cell types. PCSK9 binds to the receptor for low-density lipoprotein particles (LDL), which typically transport 3,000 to 6,000 fat molecules (including cholesterol) per particle, within extracellular fluid. The LDL receptor (LDLR), on liver and other cell membranes, binds and initiates ingestion of LDL-particles from extracellular fluid into cells, thus reducing LDL particle concentrations. If PCSK9 is blocked, more LDLRs are recycled and are present on the surface of cells to remove LDL-particles from the extracellular fluid. Therefore, blocking PCSK9 can lower blood LDL-particle concentrations. Through the inhibition of PCSK9, these novel drugs significantly reduce LDL cholesterol levels when added to statin therapy. PCSK9 levels are linearly associated with necrotic core size of plaques in nonculprit coronary arteries. The impact of PCSK9 inhibition on plaque vulnerability remains to be elucidated, but 1 PCSK inhibtor, alirocumab, is reported to reduce plaque macrophage and necrotic core content in a mouse model of atherosclerosis. The recently published GLAGOV trial (Global Assessment of Plaque Regression With a PCSK9 Antibody as Measured by Intravascular Ultrasound) reported a reduction in IVUS-derived plaque atheroma volume in patients treated with evolocumab for 78 weeks. In a post hoc analysis of the ODYSSEY LONG TERM study (Long-term Safety and Tolerability of SAR236553 [REGN727] in High Cardiovascular Risk Patients With Hypercholesterolemia Not Adequately Controlled With Their Lipid Modifying Therapy: A Randomized, Double-Blind, Placebo-Controlled Study), addition of a PCSK9 inhibitor to statin therapy resulted in better MACE-free survival (defined as death from coronary heart disease, nonfatal MI, ischemic stroke, or unstable angina requiring hospitalization). Larger prospective clinical trials with hard clinical end points are currently enrolling and will provide more information on the added value of proprotein convertase subtilisin–kexin type 9 inhibition.
______
______
New drug targets of atherosclerosis:
The lipid abnormality is one of the major modifiable risk factors for atherosclerosis. Both genetic and environmental components are associated with the development of atherosclerotic plaques. Immune and inflammatory mediators have a complex role in the initiation and progression of atherosclerosis. Understanding of all these processes will help to invent a range of new biomarkers and novel treatment modalities targeting various cellular events in acute and chronic inflammation that are accountable for atherosclerosis. Several biochemical pathways, receptors and enzymes are involved in the development of atherosclerosis that would be possible targets for improving strategies for disease diagnosis and management. Earlier anti-inflammatory or lipid-lowering treatments could be useful for alleviating morbidity and mortality of atherosclerotic cardiovascular diseases. However, novel drug targets like endoglin receptor, PPARα, squalene synthase, thyroid hormone analogues, scavenger receptor and thyroid hormone analogues are more powerful to control the process of atherosclerosis.
_
All the new drug targets and their possible mechanisms of action for the management of atherosclerosis are summarized in table below:
Drug targets and its mechanism of action for atherosclerosis.
| Drug targets | Mechanism of action |
| PPARγ selective modulator | Decreases insulin resistance |
| Anti-inflammatory effects in macrophage foam cells | |
| Endoglin receptor modulator | Reduces expression of inflammatory cell adhesion molecules |
| Increases vascular smooth muscle cell | |
| Stabilize and reduce prothrombogenic state | |
| CETP inhibitor | Increases HDL cholesterol levels |
| Reverses cholesterol transport system | |
| ACAT enzyme inhibitor | Prevents transformation of macrophages into foam cells |
| Decreases synthesis of lipoproteins | |
| PPARα agonist | Decreases TG-rich lipoproteins |
| Increases HDL | |
| Anti-inflammatory effects in vessel wall | |
| DGAT enzyme inhibitor | Inhibit transfer of acyl group from acyl-coenzyme-A to the sn-3 position of 1,2-diacylglycerol |
| Reduces triacylglycerol formation | |
| MTTP protein inhibitor | Prevents lipid assembly, transport, secretion of lipoproteins, triglyceride rich chylomicrons (in enterocytes) and VLDL |
| Modulates transportation of TG, cholesteryl ester, and phosphatidylcholine | |
| Squalene synthase inhibitor | Prevent biosynthesis of the sterol nucleus of cholesterol |
| Thyroid hormone analogues | Increases activity of lipoprotein lipase |
| Accelerates LDL-c clearance | |
| Lanosterol synthase inhibitor | Restrain four-ringed sterol intermediate formation |
| Prevent deposition of cholesterol within macrophages | |
| Cytochrome P450 modulator | Regulate conversion of cholesterol to 7α-hydroxycholesterol |
| AMPK activator | Regulates glucose metabolism |
| Decreases hepatic lipogenesis, synthesis of new FA and cholesterol | |
| Increases FA oxidation | |
| Omega-3 FAs | Inhibit expression of SREBP-1 |
| Reduces TG levels and atherogenic lipoproteins | |
| Heat shock protein-65 and CETP vaccine | Reduction in immune tolerance against self-antigen |
| Prevent transfer of CE into triglyceride-rich lipoproteins or LDL | |
| sPLA2 inhibitor | Prevents inflammatory, autoimmune and allergic reactions |
| Lp-PLA2 inhibitor | Restrain formation of LysoPC and oxNEFA |
| Reduces proinflammatory and cytotoxic products | |
| Scavenger receptor inhibitor | Inhibits macrophage activation, lipid metabolism, and inflammation |
| Prevent induction of apoptosis, apoptotic cell clearance, and pathogen recognition |
_____
Vitamin K-Antagonists Accelerate Atherosclerotic Calcification and Induce a Vulnerable Plaque Phenotype, a 2012 study:
Vitamin K-antagonists (VKA) are treatment of choice and standard care for patients with venous thrombosis and thromboembolic risk. In experimental animal models as well as humans, VKA have been shown to promote medial elastocalcinosis. As vascular calcification is considered an independent risk factor for plaque instability, authors here investigated the effect of VKA on coronary calcification in patients and on calcification of atherosclerotic plaques in the ApoE−/− model of atherosclerosis. This study shows that VKA use is associated with coronary artery plaque calcification in patients with suspected CAD and causes changes in plaque morphology with features of plaque vulnerability in ApoE−/− mice. Study findings underscore the need for alternative anticoagulants that do not interfere with the vitamin K cycle.
Authors conclude that VKA ignite a cascade of responses leading to progressive calcification and destabilization of atherosclerotic plaques. Although the use of VKA thus may impart a risk factor for acute coronary events, the relatively safe historical profile of VKA suggests, however, otherwise. Detrimental effects of VKA could well be masked by their potent inhibitory effects on the coagulant system, an important determinant in atherothrombosis. Nevertheless authors’ findings support the growing need for alternative anticoagulants and underscore a need for anticoagulants that do not interfere with the vitamin K-cycle.
_____
Oxidized LDL antibodies in treatment and risk assessment of atherosclerosis and associated cardiovascular disease, a 2007 study:
Immune responses against oxidized forms of LDL play a critical role in activation and regulation of the inflammatory process that characterizes all stages of atherosclerosis. In humans oxidized LDL is targeted by both IgM and IgG autoantibodies. Immunization of hypercholesterolemic animals with oxidized LDL has been shown to inhibit atherosclerosis demonstrating that at least some of these immune responses have a protective effect. The identification of the structures in oxidized LDL that are responsible for activation of immunity has made it possible to develop novel therapeutic approaches for treatment of atherosclerosis based on active (vaccines) and passive (antibodies) immunization. Studies performed in atherosclerosis-prone mice demonstrate that both peptide-based vaccines and recombinant IgG targeting epitopes in oxidized LDL significantly reduce atherosclerosis. There is also evidence that antibodies against oxidized LDL could also be used for imaging atherosclerosis.
Note:
A relationship between circulating antibodies against Ox-LDL and atherosclerotic disease has not been unequivocally shown.
______
______
Vulnerable plaque controversies:
_
Limitations to the Vulnerable Plaque Paradigm:
From the view of vulnerable plaque detection, morphological studies should provide valuable insight regarding which criteria are important for localizing high-risk lesions using imaging modalities and assessing the potential risk for rupture. Obvious important cause-and-effect data are missing from the current paradigm because of our inability to accurately detect vulnerable plaques in humans in vivo and because of the lack of an animal model. In this regard, representative lesions in current animal models rarely progress beyond the stage of fibroatheroma; more often, they consist of only masses of lipid-laden intimal macrophages without a well-developed fibrous cap or necrosis. Lesions with this histology rarely become clinically significant except in examples of severe hyperlipidemia, in which the lumen can become obstructed by the sheer plaque burden, a situation that is quite atypical in human disease. Therefore, a major limitation today exists partly because the precise mechanisms of progression from an asymptomatic stable to high-risk plaque (TCFA or vulnerable plaque) that lead to rupture and thrombosis are incompletely understood.
Although mouse models of atherosclerosis have provided important insights into the genetic component of the disease, major differences in the pathways directed toward lesion composition between mice and humans remain apparent. For example, inflammation is the most abundant plaque component in mice, whereas in humans it constitutes only 2% to 5% of total lesion volume. Moreover, intraplaque hemorrhage from leaky vasa vasorum in human plaques is believed to be a significant factor in necrotic core expansion, a phenomenon rarely observed in mice. Perhaps more importantly, the mechanisms of disease initiation and progression are quite distinct, considering that mouse lesions are products of macrophages, whereas in humans, early lesion progression arises from smooth muscle cells enriched in an environment of proteoglycans and collagen with discreet pools of lipid, so-called pathological intimal thickening. Although features of fibrous cap formation, protease, activation, and cell death in mice are reminiscent of human plaques, the applicability of these to the natural progression of the disease has not yet been tested.
The merit of imaging “vulnerable atherosclerotic plaques” remains highly controversial. Results from optical coherence tomography and intravascular ultrasound imaging in patients with coronary heart disease suggest that certain individual coronary atherosclerotic plaque characteristics, e.g., large lipid core in a fibroatheroma, are associated with greater risk of adverse patient outcome. However, a closer look at these studies reveals that these associations are confounded by the relationship of “vulnerable plaque” characteristics with baseline lumen obstruction, which is a known predictor of recurrent angina and the main component of the reported adverse patient outcome. Recent insights into the pathophysiology of acute coronary syndromes suggest it to be an exceedingly complex process involving numerous local and systemic factors, which hinders outcome prediction. The quest for the vulnerable plaque rests on the erroneous assumption that detecting coronary atherosclerotic lesions, which are prone to rupture or erode, will identify individuals at high risk of suffering acute coronary events. However, there is strong and consistent evidence suggesting that plaques most commonly rupture without associated clinical symptoms. Instead, ruptured plaques typically heal clinically silently and lead to plaque progression. The atherosclerotic disease burden, its metabolic activity, and risk factors for an inadequate response by the coagulation system to plaque disruption, on the other hand, are important predictors of acute coronary event risk and deserve our attention more than individual plaques.
____
____
Challenges to the Vulnerable Plaque Concept:
The concept of visualization and treatment of the vulnerable plaque has predominantly focused on plaque rupture, as this is the pathophysiological mechanism most frequently responsible for ACS. A review of literature, which included 22 autopsy studies and a total of 1847 coronary arteries, reported that plaque rupture is observed in 73% of all fatal coronary thrombi. However, contemporary optical coherence tomography studies have provided preliminary evidence that the prevalence of plaque rupture in nonfatal ACS, and especially in non–ST-segment–elevation myocardial infarction (non-STEMI), might be lower. The prevalence of plaque rupture in these studies was reported to be 44% to 73%, whereas plaque erosion and calcified nodule were reported to be only 27% to 33% and 8%, respectively. Although plaque rupture remains the most prominent cause of ACS, it should be noted that novel therapeutics aimed at preventing plaque rupture may have no effect on the incidence of ACS as a result of plaque erosion or calcified nodule.
Another challenge to the concept is the fact that not all cases of intracoronary thrombosis necessarily lead to a clinical event. A recent review by Arbab-Zadeh et al examined the results of many clinical and pathological investigations and reported subclinical plaque rupture in 11.5% of patients with stable CAD and 21.5% in patients who presented with ACS. Subclinical plaque ruptures, however, are not without consequences, as they are considered an important mechanism responsible for increased plaque burden and subsequent lumen narrowing. In line with this hypothesis, Park et al have recently shown that several suspected coronary computed tomographic angiography (CCTA)–derived vulnerable plaque features are correlated with ischemia-causing lesions as defined by an abnormal fractional flow reserve.
The focus on imaging and treatment of specific vulnerable plaques fails to address the fact that atherosclerosis is a systemic, multifactorial disease, and optimal care likely consists of systemic and multifocal diagnosis and therapy, of which local plaque therapy may be a component. In addition, knowledge on the prevalence and natural history of suspected imaging-derived vulnerable features is limited. Longitudinal imaging studies have suggested that plaque morphology may change over time, and many suspected vulnerable plaques may not progress over the course of years and may even regress to more stable lesions. Actually, the vast majority of thin-capped, lipid-rich atheroma persists for years without causing any clinical event. Although the identification of rupture prone plaques in vivo will certainly not be able to predict the occurrence of all coronary events, it could provide additional prognostic information to target novel therapeutics. Altogether, lack of clinical outcome studies proving that a suspected vulnerable plaque actually causes a coronary event hampers the clinical applicability of the concept.
Another important topic to notice is that thin-capped, lipid-rich atheroma are not always solitary, but are often multiple, and affect several arterial beds in the same individual. Besides all this, there is not an excellent clinical or anatomical feature that will predict which of these coronary plaques may rupture or erode. In the Providing Regional Observations to Study Predictors of Events in the Coronary Tree (PROSPECT) study, only approximately 5% of thin-capped plaques as defined by virtual histology caused coronary events during a 3.4-year follow-up period. As longitudinal intravascular imaging studies such as PROSPECT enrolled higher risk patients, thin-capped plaques in lower risk populations may cause even fewer thrombotic events. Thus, the vast majority of so-called ‘vulnerable plaques’ does not exhibit clinical ‘instability’ and indeed seldom provokes ACS. Moreover, the consequences of a plaque disruption depend not only on the ‘solid state’ of the atheroma itself, but also on the fluid phase of blood, for example the concentrations of fibrinogen, endogenous inhibitors of fibrinolysis, and pro-coagulant micro particles.
Thus, besides all this development in the capacity of detecting these dangerous spots in the coronary arteries, there is currently no significant evidence that treating these vessels locally (with coronary stenting, for example) could protect the patient from having an ACS. In other words, there is no evidence that finding a vulnerable plaque and treating it locally can lead to improved outcomes. In contrast, systemic therapies, with oral drugs like statins and antiplatelet, apparently can greatly improve outcomes in patients with vulnerable plaques. Identifying vulnerable patients (not only vulnerable plaques) and treating them aggressively, seems to bring more benefits than invasively investigating and treating vulnerable plaques locally. Besides, it is easier and cheaper to identify vulnerable patients than vulnerable plaques.
______
______
Calcium contradiction:
Rupture of atherosclerotic plaque is a major cause of human mortality worldwide, which makes the prerupture identification of vulnerable atheroma extremely important for patient risk evaluation. Evidences have shown that the composition of an atherosclerotic plaque, rather than its degree of stenosis or size, is usually of more importance for acute clinical events. Generally a vulnerable plaque is often found to be associated with a thin fibrous cap, a high inflammation burden, a large lipid pool, macroscopic heterogeneity, and so on. Calcification is commonly believed to be associated with cardiovascular disease burden. Recently, the influence of calcification on plaque vulnerability has raised many research interests. There are many ways to image the calcification in plaque, such as noninvasive molecular imaging probes. Chen and Dilsizian used the molecular probe 18 F-sodium fluoride (18 F-NaF) for positron emission tomography (PET) imaging, which targets active microcalcifications in atherosclerotic plaques. Kimura et al. revealed a significantly higher frequency of lipid-rich plaque with microcalcification in lesions with echo signal attenuation.
_
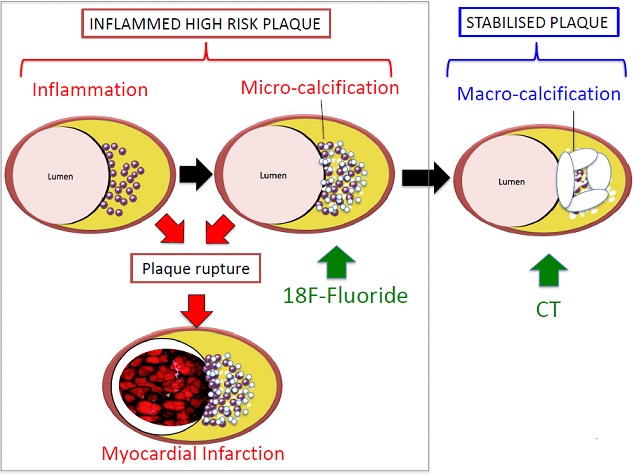
Figure above shows demonstration of micro-calcification and macro-calcification by 18F-NaF and CT respectively.
_
Studies from Mauriello et al. showed that the calcification, as well as its distance from the lumen, is not correlated with the presence of unstable plaques. Thus, the authors suggested that the calcification is not useful to identify the vulnerable plaque. Hermann et al. found that individuals suffering a stroke have significantly higher coronary artery calcification (CAC) values at baseline than the remaining individuals, and furthermore CAC is an independent stroke predictor in addition to classical risk factors for those patients at low or intermediate vascular risk. Moreover, mechanical experiments on human carotid plaques by Mulvihill et al. showed that calcification in the tissue structure may lead to increased vulnerability of the plaque. On the other hand, it was demonstrated by Shaalan et al. that symptomatic plaques are less calcified and more inflamed than asymptomatic plaques, implying that the calcification may reduce the plaque rupture risk. Wahlgren et al. investigated thirty carotid endarterectomy plaques which were classified as noncalcified and calcified, and obtained a similar result that fibrous cap inflammation is more likely to occur in noncalcified than in calcified plaques, suggesting that plaque calcification may result in protection against the rupture of plaque.
_
Computational studies on microcalcifications have also been investigated previously. Kelly-Arnold et al. examined the spatial distribution, clustering, and the shape of different microcalcification size in fibrous caps and found that nearly all fibrous caps have microcalcifications, but only a small subset has rupture potential. Bluestein et al. developed a fluid-structure interaction (FSI) model to study the microcalcification effects on the plaque vulnerability and found that calcification can increase plaque vulnerability. Cilla et al. investigated the effect of microcalcifications on the stress field of an atheroma plaque vessel section by performing a parametric finite element study on an idealized model. Vengrenyuk et al. investigated the stress distribution using the multilevel micro-CT based 3D numerical modeling techniques. Results showed that the peak circumferential stress increases with the existence of calcifications (inclusions) and may grow even higher by elongated microcalcifications, while in contrast, macrocalcifications in cap shoulders were shown to actually increase the plaque stability.
__
Hard calcified plaque least vulnerable: American journal of cardiology 2011 article:
One major advantage that coronary CT angiography has over cardiac calcium scoring is that the former can distinguish whether areas of plaque in the coronary arteries are hard or soft. A coronary CT angiogram can distinguish between these types of plaque, while the coronary calcium score cannot. Plaque contents, it’s distribution and consistency make it vulnerable. Soft spots formed by lipids may result in plaque cracks and fissure. Semi solid, mixed, gel like soft plaques are dangerously prone for rupture. Oxidation of LDL, LDL liquefaction and tissue metalloproteinase, thickness of fibrin caps , all promote softening. If none of above mechanism is operative in a given patient, the plaque becomes stiff and hard. Calcification is the ultimate in hardening. Calcified plaque is resistant to mechanical deformation. If stiff plaques are less vulnerable, hard plaques (i.e. calcified plaques) must be least vulnerable. Calcification can be called an end result of coronary atherosclerosis. So, calcified coronary artery can be referred to as a failed mission of atherosclerosis. It is equivalent to death of atherosclerosis and denotes the end process of this dreaded disease process.
_
Hard, not soft, plaque buildup may predict risk of heart attack, a 2017 study:
Previous research has suggested that it is the soft – that is, noncalcified and lipid-laden – plaque that holds the highest risk of rupturing and triggering heart attacks. However, new research – presented at the American College of Cardiology Scientific Sessions, in Washington, D.C. – indicates that the opposite may be true. The new study, conducted by researchers from the Intermountain Medical Center Heart Institute in Salt Lake City, UT, suggests that hard, calcified plaque may be a stronger indicator of adverse cardiovascular events.
The scientists analyzed the composition of coronary artery plaque in 224 patients who had diabetes but who showed no signs of heart disease. The patients were registered with the Intermountain Medical Center Heart Institute, and the data were collected as part of a previous study carried out in collaboration with the Johns Hopkins School of Medicine and National Institutes of Health (NIH). The new research focused on the long-term associations between plaque buildup and cardiovascular disease, as the scientists followed the patients for almost 7 years, on average. To analyze the composition of the plaque, the researchers used a coronary angiography performed with computed tomography (CT). The CT scan was stratified into layers of noncalcified, calcified, and fibrous plaque. The researchers correlated the data with the calculated risk of unstable angina (a condition in which the heart does not receive enough oxygen-rich blood), heart attack, and death. The study revealed that higher amounts of calcified plaque strongly predicted serious coronary conditions. “It is a disease marker, not a risk marker. And we think it is possibly a very important predictor,” says Dr. Brent Muhlestein, one of the study’s authors and co-director of cardiology research at the Intermountain Medical Center Heart Institute. Dr. Muhlestein also explains the importance of the findings for heart disease diagnosis and treatment: ” The finding potentially could mean a lot of patients may not require statin therapy, even though they have high cholesterol. Maybe we can find and identify them. If there is no atherosclerosis, you are not going to have a heart attack. So the coronary calcium score may allow us to much more effectively select who we treat.”
Although the calcium deposits cannot be completely removed from the arteries, aggressive statin therapy can improve the health outcomes for patients with atherosclerosis. Additionally, the new findings may change the way that physicians determine who is at risk of heart attack or heart failure. Dr. Muhlestein notes that more studies are needed to confirm their findings, as well as to understand the mechanism behind this correlation. “We need further validation to gauge the importance of why the coronary calcium score is so predictive,” he says.
_
Co-localization of plaque macrophages with calcification is associated with a more vulnerable plaque phenotype and a greater calcification burden in coronary target segments as determined by OCT, a 2018 study:
Plaque macrophages reflect plaque inflammation and play a role in lipid accumulation as well as in the disruption of the fibrous components of the plaque inducing a more vulnerable plaque phenotype prone to plaque rupture. On the other hand, both optical coherence tomography (OCT) and intravascular ultrasound (IVUS) have been able to identify small calcifications as features of plaque vulnerability; in particular, Ehara et al. found spotty calcifications, i.e. calcifications with a calcium arc<90° to be more frequently present in culprit lesions of ACS patients rather than in lesions of patients with stable CAD. These findings could be confirmed using the superior resolution of OCT. Another interesting aspect of the relationship between calcifications and plaque vulnerability was the hypothesis that the so called microcalcifications may increase the peak circumferential stress of the fibrous cap, thus potentially promoting plaque rupture and triggering ACS. Interestingly, data from basic science studies suggest that calcifications and macrophages are interconnected: microcalcifications, for instance, promote inflammation in vitro and—vice versa—macrophages foster microcalcifications, e.g. via microvesicles and induction of an osteogenic phenotype in vascular smooth muscle cells. Despite the known interaction between macrophages and calcifications in vitro, there is no clinical data investigating the co-localization between plaque macrophages and calcifications (ColocCaMa) in coronary arteries in living patients. The use of OCT enables in vivo visualization of both, macrophages and calcifications in coronary arteries. Given the known reciprocal interaction between plaque macrophages and calcifications in vitro, a new study aimed to quantify the ColocCaMa in the coronary target segments and its implications towards further features of plaque composition and plaque vulnerability using OCT. The study found that plaque macrophages co-localize with calcifications in coronary target segments and this is associated with high-risk morphological features including microcalcifications and macrophage infiltration as well as with greater calcification burden.
__
In a nutshell:
The role that the calcification plays in plaque vulnerability is still under debate. Some studies indicated beneficial effects in stabilizing the plaque, making it stiffer and less prone to rupture, while others tended to believe it would increase the risk of plaque rupture. Microcalcification within the thin fibrous cap, or spotty calcification, may serve as a marker of plaque vulnerability. In contrast, larger calcified sheets consisting of merged areas of microcalcification are associated with plaque stability. Previous research has suggested that it is the soft – that is, noncalcified and lipid-laden – plaque that holds the highest risk of rupturing and triggering heart attacks. However, new research suggests that hard, calcified plaque may be a stronger indicator of adverse cardiovascular events. Balance of arguments suggests that CAC is a disease marker and correlated with adverse cardiovascular events no matter soft or hard plaque, no matter microcalcification or macrocalcification.
______
______
Questioning the Vulnerable Plaque Hypothesis:
Considerable issues are currently being raised with regard to the vulnerable plaque hypothesis context, especially its clinical significance. Despite vulnerable plaque features presence having high sensitivity in cases of acute events, prospective studies, such as Providing Regional Observations to Study Predictors of Events in the Coronary Tree (PROSPECT), VH‐IVUS in Vulnerable Atherosclerosis (VIVA), and the European Collaborative Project on Inflammation and Vascular Wall Remodeling in Atherosclerosis (ATHEROREMO‐IVUS), have consistently shown that their positive predictive value is low, implying low prognostic value on an individual basis, used in addition to classical risk‐stratification factors. In the same framework, the PREDICTION study only demonstrated an ability for IVUS and shear stress features to predict plaque enlargement and lumen narrowing, not acute events. Failure of a shear‐stress inclusive approach to define plaque vulnerability may be related to IVUS resolution, considerably inferior to that of OCT. Moreover, in the recently presented 2‐year follow‐up of the COLOR registry, NIRS‐determined culprit plaque vulnerable phenotype was not associated with a difference in major adverse cardiovascular events occurrence (5.4% vs 6.3% in the nonvulnerable plaques; P=0.41), suggesting that a stented lesion has the same possibility to cause a new clinical event regardless of its type. In fact, adverse events during follow‐up were more commonly related to nonculprit lesions; however, outcomes for the NIRS‐defined vulnerable nonculprit plaques have not yet been analyzed. In conjunction, the full results from the COLOR and LRP studies are expected to definitely answer whether NIRS fares any better in identifying plaques prone to causing future events.
Recent findings appear to suggest that the notion of nonstenotic plaques underlying ACSs is a misconception, given that, when having been assessed close to the episode of which they were the culprit, plaques appear severely stenotic, having undergone a period of accelerated growth. Although vulnerable plaques may carry high lipid burdens, masked by their expansive remodeling, one of the major tenets of the vulnerable plaque hypothesis is that stenosis degree has little impact on the plaque’s fate, compared to its structural features (although nothing actually precludes it from being severely stenotic). However, this controversy can be reconciled by considering that plaques with accelerated progression are, in fact, vulnerable plaques, fueled by repeated episodes of hemorrhage and subclinical (healed) rupture and erosion.
Studies suggesting plaques may alternate between vulnerable and nonvulnerable phenotypes cast doubt on the significance of diagnosing plaque vulnerability and, conversely, suggest that nonvulnerable plaques may transition into an unstable morphology and suffer rupture or erosion. The stochastic (by current knowledge), rather than determinate, nature of plaque progression is indirectly supported by the finding that extensive nonobstructive atherosclerosis confers similar risk for MI and sudden death as obstructive, but less‐extensive, disease. Additionally, occurrence of clinical events heavily depends on blood and myocardial features (as seen in the figure below); thus, significance of treating rupture prone plaques becomes unclear. However, it is not inconceivable that plaque interdependence may extend well beyond an observed domino‐like effect (a destabilized plaque releasing mediators knocking others—even in remote vascular beds—out of balance). More specifically, flow alterations following a silent event or even a successful coronary intervention could inevitably, and currently unpredictably, alter the course of even distant lesions. Finally, the number of nonculprit, asymptomatic, potentially type‐switching plaques may be such, especially in cases of high atherosclerotic burden that renders, in combination with the above, all efforts at localized “vulnerability‐guided” treatment, without the ability to accurately assess implications futile, if not dangerous.
_
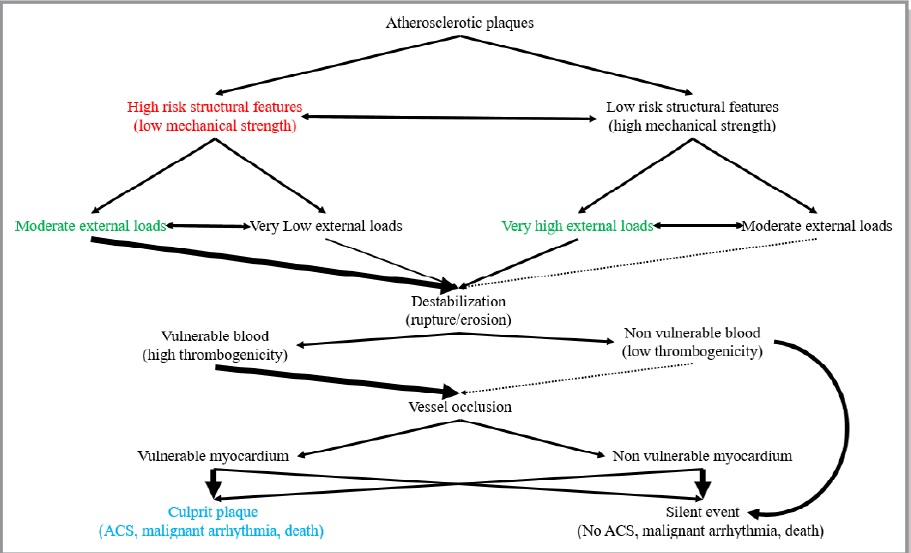
Figure above shows projected natural course of atherosclerotic plaques based on their intrinsic and extrinsic features. This figure depicts potential evolution of plaques based on an integrative assessment, including external and internal factors, and underlies the necessary paradigm shift in the vulnerable plaque definition. Size and thickness of arrows implies relative probability for a course between the two listed in every step. Obviously, the default state, that is, maintenance of the status quo with plaques remaining quiescent, is far more likely in all cases—no arrow denotes certainty for an event. As seen in the diagram, even stable, by all accounts, plaques can rupture/erode. This can be attributed to a phenotype/external stressor shift, an erroneous assessment, or simply an unlikely event given that plaque behavior is, as everything in medicine, the sum of probabilistic, not determinate, events. Additional levels of uncertainty are inserted through the, currently unpredictable, blood and myocardium response to a destabilizing event. Nonvulnerable blood and plaque destabilization combination may also lead to events, albeit rarely. “Silent” events are considered, for the purposes of this figure, both those resolving previous to thrombus formation and those with thrombosis yet remaining clinically undetected. “High” and “low” risk structural features can only be defined relatively, that is, how would the plaque behave in cases of applied loads that can be considered moderate. Obviously, further research is necessary to elucidate these parameters. Color coding: blue—initial definition; red—current perception; green—integrative approach. Double‐headed arrows denote bidirectional processes. ACS indicates acute coronary syndrome.
_
After 20 years of research and countless clinical studies, has any method of detection of high-risk plaques successfully predicted which lesions are most likely to rupture, resulting in an acute event? Astonishingly, the answer is clearly “no.” There exist no prospective clinical trials demonstrating that presence of any specific plaque morphological feature is associated with a worse prognosis for patients. While it is true that autopsy studies suggest that TCFAs are most often the culprit lesions in acute coronary syndromes (ACS), it is quite another matter to show that the presence of this morphology prospectively predicts plaque behavior. In the absence of a proven relationship between TCFAs and adverse outcomes, improved methods for detection of such lesions constitute only an academic exercise.
_
Regarding treatment options, it is true that numerous vulnerable plaques usually coexist in patients at risk for cardiovascular events. This, in combination with the domino effect during an ACS, potential for plaque morphology alteration with regression of the vulnerable phenotype and presence of further determinants of clinical outcome (vulnerable blood—vulnerable myocardium) argue against isolating and locally treating vulnerable plaques, and in favor for implementing systemic atherosclerosis treatment approaches, interpreting presence of these plaques to signify the degree of its progression in terms of natural history. Exceptions to the above may arise in cases of nonculprit destabilized plaque detection during an acute event (potential for domino effect, as suggested by findings of the CvLPRIT [Complete versus Lesion‐only Primary PCI trial] trial), presence of accelerated plaque progression with increasing stenosis (nonetheless requiring close patient follow‐up), and establishment of clinically applicable criteria for imminent plaque destabilization. Thus, in this aspect, more judicious use of targeted localized therapies is warranted, as indeed suggested by vulnerable plaque skeptics.
______
______
The Myth of the “Vulnerable Plaque”:
Transitioning from a Focus on Individual Lesions to Atherosclerotic Disease Burden for Coronary Artery Disease Risk Assessment, a 2015 review:
Numerous factors (e.g., dyslipidemia, diabetes, and others), are associated with increased rates of adverse events; however, their hazard rates are too small for accurate individual risk assessments. The DIAD (Detection of Ischemia in Asymptomatic Diabetics) trial revealed that after 4.8 years of follow-up, 97% of asymptomatic patients with diabetes mellitus remained free of myocardial infarction and cardiac death. Even when combined as comprehensive risk scores (e.g., by the Framingham study), predictive accuracy is insufficient for adequate individual risk assessments, leading to substantial overtreatment and undertreatment, with associated morbidity and societal costs. The mechanisms leading to adverse events from atherosclerotic disease are clearly more complex than initially assumed, explaining our difficulties in accurately predicting events in individuals. In addition to the presence, extent, and metabolic activity of atherosclerotic disease, individual adaptations and responses to thrombogenic stimulation from altered vascular function are critical for determining the risk of acute coronary events. Despite a consensus on the complexity of acute coronary event risk evaluation and the necessity for comprehensive patient assessment, recent efforts to identify high-risk patients have focused on using advanced imaging methods to detect single “vulnerable” atherosclerotic plaques. This narrow focus neglects the complexity of the processes leading to risk and lacks supporting evidence.
_
The “Vulnerable Plaque” Concept:
Pathology studies have demonstrated the common association of acute myocardial infarction with the rupture or erosion of a coronary atherosclerotic plaque, most frequently a thin-cap fibroatheroma (TCFA), characterized by a large lipid or necrotic core separated from the coronary arterial lumen by a thin membrane cap. Thus, the identification of TCFAs in humans was assumed to signify a high risk of ensuing acute coronary events, which then might necessitate directed treatment or specific preventative measures. Accordingly, enormous efforts have been undertaken to enable the identification of TCFAs and other high-risk plaque features in humans.
_
Limitations of Studies Supporting the High-Risk Atherosclerotic Plaque Concept:
A number of clinical investigations used various imaging tools to identify high-risk atherosclerotic plaque features in order to predict an increased risk of adverse events. In one large, prospective clinical study, PROSPECT (Providing Regional Observations to Study Predictors of Events in the Coronary Tree), rates of adverse cardiac events according to types of coronary atherosclerotic plaque were investigated in more than 600 high-risk patients studied with IVUS-virtual histology. Although 596 TCFAs were identified, only repeat hospitalization for chest pain was associated with events. This was expected, given the typically larger luminal encroachment of TCFAs compared with pathological intimal thickening (the prevalent type of lesion in the study). However, the risk of myocardial infarction or sudden cardiac death related to these lesions was very low (see figure below). A similar study using IVUS-virtual histology (VIVA [VH-IVUS in Vulnerable Atherosclerosis]) reported nearly identical findings. Studies using OCT revealed greatly detailed plaque characteristics in patients with acute coronary syndromes and other at-risk populations. Similar to the information provided by IVUS, studies using OCT suggest that a larger lesion plaque burden might indicate an increased risk of acute coronary events. Noninvasive imaging studies of the coronary arteries using computed tomographic angiography reported increased rates of acute coronary syndromes in patients with low-attenuation plaques (presumably high in lipid content) with external remodeling compared with those without such plaques. However, all of these studies claiming independent risk prediction of certain plaque features share the fundamental limitation that the atherosclerotic disease burden was not considered as a potential confounder. These “high-risk” features are conceivably mere markers of more extensive and/or active atherosclerotic disease compared with the control group. Given the overwhelming evidence for disease burden as a powerful predictor of outcome, any additional risk features should be assessed against it before we assume independent risk prediction. Therefore, despite promising results from a number of clinical studies, there is no conclusive evidence for truly independent risk prediction associated with high-risk plaque features. High-risk features may still be valuable as markers for disease burden or activity; however, such value has not been established.
_
Risk of MI or Death Associated with Individual Plaques in the PROSPECT Study:
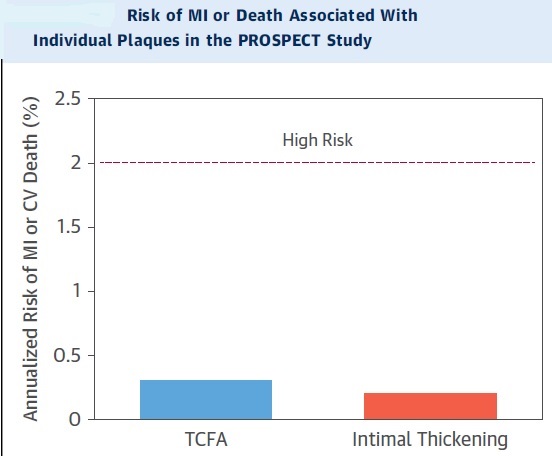
Figure above shows maximum annualized risks (percent) of myocardial infarction (MI) or cardiovascular (CV) death associated with individual coronary atherosclerotic plaques identified at baseline by intravascular ultrasound-virtual histology in the PROSPECT (Providing Regional Observations to Study Predictors of Events in the Coronary Tree) study are shown. The rates are based on the occurrence of 6 events (6 myocardial infarctions and no deaths) after 3.4 years of follow-up among 1,005 coronary arterial sites with pathological intimal thickening and 595 thin-cap atheroma (TCFAs), assuming all events were caused by the respective plaque type, thus representing the worst-case scenario. Considering equal risk among the 3,160 plaques detected at baseline (best-case scenario), the event rate associated with each plaque would be only 0.06% per year. The risk of MI or death associated with individual TCFAs or vulnerable plaques is much smaller than what is conventionally considered high risk, even when maximal risk is assumed.
_
Evidence Diminishing the Significance of Identifying “High-Risk” Plaques:
Despite our ability to identify atherosclerotic lesions that exhibit vulnerable characteristics using various imaging tools, clinical studies have failed to demonstrate meaningful clinical utility for plaque imaging. These negative results are explained by the numerous pathological and clinical investigations demonstrating that many (if not most) plaques rupture without clinical syndromes. The percent of patients with subclinical plaque ruptures varies with their risk profile and the sensitivity of assessment methods, ranging from 4% to 79% (see figure below). Plaque rupture and its healing are frequently clinically silent but lead to progressive lumen obstruction. In lesions with advanced lumen narrowing (>50% stenosis), histopathology almost invariably reveals 1 or more healed subclinical plaque ruptures. Because healed plaque ruptures can be detected by pathology or imaging only within a certain time frame after the initial plaque disruption, the true rate of asymptomatic plaque ruptures is probably underestimated. In most patients with advanced atheroma, plaque rupture and subsequent healing have already occurred. Pathology studies show that approximately 10% of the subclinical United States adult population exhibits advanced coronary atheroma, so it is reasonable to assume that many millions of persons unknowingly experience plaque ruptures each year.
_
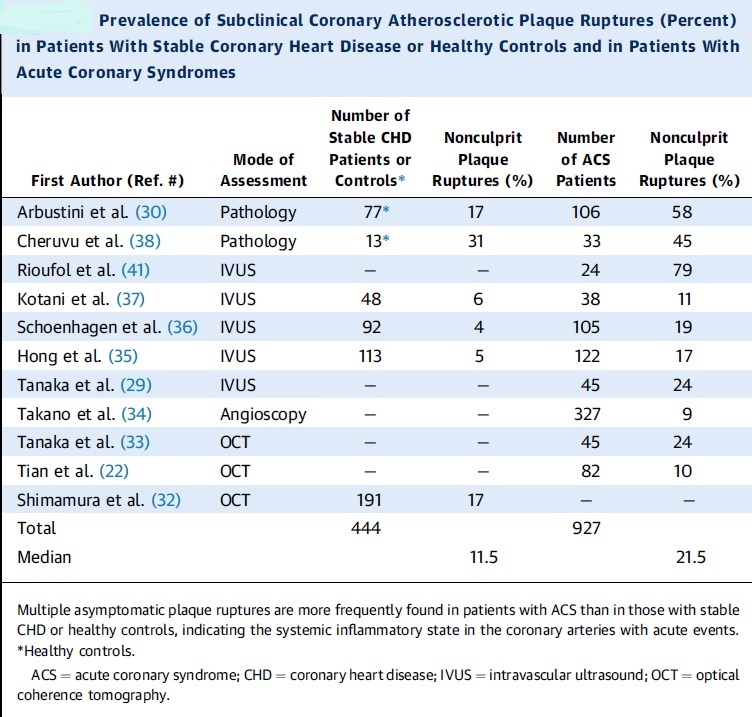
Figure above shows prevalence of Subclinical Coronary Atherosclerotic Plaque Ruptures (Percent) in Patients with Stable Coronary Heart Disease or Healthy Controls and in Patients with Acute Coronary Syndromes.
_
Several longitudinal imaging studies in humans have demonstrated that plaque morphology changes over a few months, gaining or losing “vulnerable” characteristics. Using IVUS, Kubo et al. found that 75% of TCFAs transition to thick-cap fibroatheromas or fibrotic plaques within a 12-month interval, presumably secondary to rupture and healing. None of these patients experienced events during this period, providing further evidence of frequent subclinical plaque alterations. A recent study used OCT to confirm the dynamic nature of coronary atherosclerotic disease, demonstrating that TCFAs in various stages of development are highly prevalent in patients with coronary atherosclerotic disease. In patients with acute coronary syndromes, plaque ruptures are frequently found apart from the culprit lesions, indicating that vulnerability is disseminated throughout the coronary tree. This suggests that detection of a state of vulnerability in a patient (e.g., widespread inflammation) is more important than detection of individual sites of vulnerability. The emerging picture of acute coronary event pathophysiology suggests a widespread, systemic condition with great unpredictability as to which particular lesion will be associated with clinically significant vascular thrombosis. Although plaque ruptures and erosions are indeed responsible for most culprit lesions in patients with acute events, because the frequency of subclinical plaque ruptures is vastly underestimated, the assumption that identifying lesions prone to rupture will prevent acute coronary events was unrealistic. Of the many plaque ruptures occurring in patients with atherosclerotic disease, very few will trigger symptomatic events, rendering it exceedingly difficult to predict adverse outcomes associated with particular lesions. Identifying a single TCFA or other “high-risk plaque,” without considering other clinical or imaging characteristics, is unlikely to be of incremental benefit for risk prediction over established factors (e.g., the extent and distribution of atherosclerotic plaque burden), because of the low risk associated with a given individual plaque and the temporal relationship of its vulnerable characteristics.
_
Changes in TCFAs During 12 Months of Follow-Up:
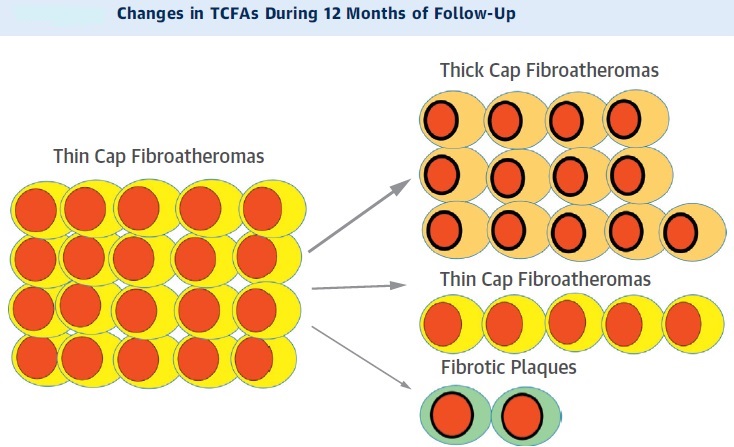
Figure above shows that changes in thin-cap fibroatheromas (TCFA) observed with intravascular ultrasound-virtual histology 12 months after baseline imaging. Of 20 TCFAs, only 5 remained unchanged, while 15 (75%) lost vulnerable characteristics and revealed thickening of the fibrous cap or transformed into fibrous plaques. The data (Kubo et al.) demonstrate high metabolic activity in atherosclerotic lesions and the short-lived nature of vulnerable plaque characteristics. None of the 99 patients experienced any events during follow-up; thus, many of the observed changes were likely the result of subclinical plaque ruptures.
_
Current Paradigm of Acute Coronary Event Pathophysiology:
Over the past few decades, clinical and laboratory investigations have led to a more complex concept of the pathophysiology of acute coronary events, involving numerous processes, many with poorly understood interactions. Although the occurrence of acute coronary events typically requires alterations of coronary atherosclerotic plaques (rupture or erosion), a thrombosis-promoting milieu is necessary to allow a clinically significant decrease in coronary blood flow and associated myocardial ischemia. Such a setting appears to result from an unfortunate constellation of prothrombotic features, for example, in patients with increased inflammatory activity and systemic or local suppression of fibrinolytic performance, an extraordinarily large stimulus for thrombosis, vasoconstriction, and/or others. The respective contributions of these factors (some hereditary, some environmental) and their temporal relationships necessary to trigger clinically meaningful vascular thrombosis are unknown. Factors favoring thrombosis need to be collectively sufficient to tilt the scale away from localized thrombus and toward extensive vascular thrombosis. See figure below. Because numerous factors influence the performance of the coagulation system at any given point in time, acute coronary events may arise as result of a “perfect storm” scenario, in which plaque disruption occurs in a specific, thrombosis-promoting setting. The risk of an acute coronary event equals the probability of plaque rupture or erosion coinciding with vascular thrombosis-promoting conditions that cannot contain the thrombus in the vascular wall. Frequent plaque ruptures, as with a large, metabolically active atherosclerotic disease burden, increase the chance that a plaque rupture coincides with a thrombosis-conducive setting. Accordingly, the strongest predictors of adverse events are the magnitude and activity of the coronary atherosclerotic plaque burden and the number of risk factors for a prothrombotic milieu, a concept supported by many clinical studies and epidemiologic data. Prothrombotic risk factors include CRP, dyslipidaemia, platelet count, homocysteine, tobacco, diabetes and genetic predisposition.
_
Fate of Ruptured Coronary Atherosclerotic Plaques According to Thrombotic Milieu:
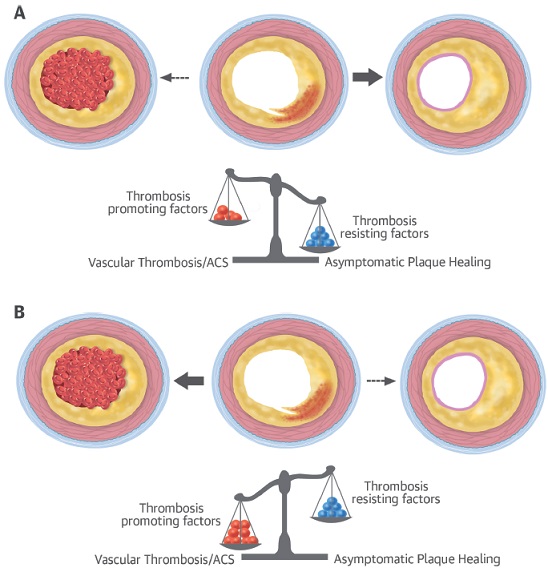
Figure above shows hypothesized interplay of prothrombotic and thrombosis resisting and containing factors that presumably determine the outcome of a ruptured coronary atherosclerotic plaque is shown.
(A) In the most common scenario, small thrombus formation associated with plaque rupture is contained and vascular occlusive thrombus is inhibited.
(B) In the less common scenario of several prothrombotic factors coinciding (e.g., inflammatory state, large lesion plaque burden, vasoconstriction, circadian rheological changes), local thrombosis associated with plaque rupture cannot be contained, and clinically significant vascular thrombosis occurs, triggering an acute coronary syndrome (ACS). The constellation of factors leading to these different outcomes is unknown.
_
Lesion Focused Versus Disease Burden Focused Risk Assessment and Management:
Numerous clinical studies using conventional invasive coronary angiography, IVUS, and cardiac computed tomography have confirmed the strong relationship between atherosclerotic disease burden and risk for adverse events. Halting coronary atherosclerotic disease progression and/or altering the vascular thrombosis-promoting milieu via platelet inhibition and risk factor interdiction are approaches proved to lower myocardial infarction and death rates. Conversely, meta-analyses have not demonstrated reduced rates of myocardial infarction or death with lesion-based treatment (i.e., percutaneous coronary intervention) compared with medical therapy in patients with stable coronary artery disease. Contradicting some earlier reports, no benefit was shown, even when selecting patients with hemodynamically significant stenoses. These results confirm that risk is most strongly conveyed by the extent of coronary artery disease, but not necessarily by individual lesions, even when highly obstructive. This supports the controversial idea that severe coronary artery stenoses confer no greater risk of triggering acute coronary events than mild lesions. Earlier angiographic studies suggested that most myocardial infarctions arise from mild coronary artery stenoses, but recent data question this paradigm. Thrombus material accumulates over several days after a plaque disruption, which may not allow lesion size and a partly organized thrombus to be accurately distinguished. Pathology studies in patients who died suddenly found culprit lesions to have an average diameter stenosis of approximately 50%, with no clear relationship between stenosis severity and risk of death. Conversely, acute coronary death rarely results from lesions with <30% luminal stenosis. Thus, a certain local plaque volume appears necessary to trigger vascular thrombosis. However, given the lack of benefit with coronary stenting, as well as the large number of obstructive lesions found by imaging and autopsy in patients without symptoms of acute coronary events, stenosis severity is unlikely to substantially alter such risk beyond a particular threshold. Patients with high-grade coronary artery stenoses may conceivably carry an increased risk of myocardial infarction and death because these lesions are markers for advanced atherosclerotic disease in the coronary tree. Consistent with this notion, nonobstructive and obstructive coronary artery disease are associated with similar risks of myocardial infarction and death if the former affects a larger number of coronary arterial segments (see figure below). Overall, strong evidence supports addressing the extent and activity of the atherosclerotic burden and thrombosis-promoting risk factors for improved patient outcomes, but there is no conclusive evidence of incremental risk reduction with lesion-specific treatment.
_
Risk Due to Nonobstructive Versus Obstructive Coronary Artery Disease:
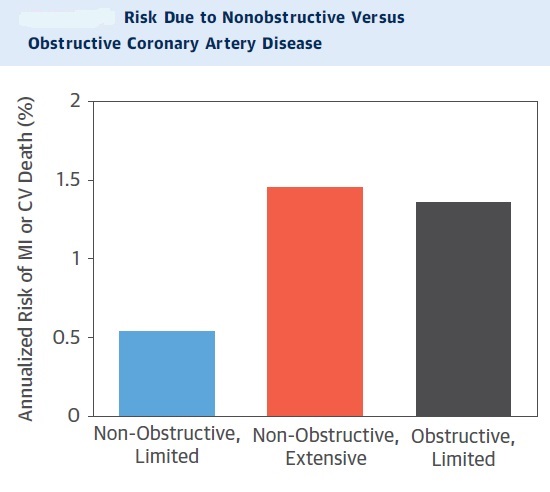
Figure above shows Annualized risk (percent) of myocardial infarction (MI) or cardiovascular (CV) death in 3,242 patients followed for a median of 3.6 years after baseline computed tomographic coronary angiography, according to the extent and severity of coronary artery disease. Risk is low in patients with nonobstructive disease (<50% stenosis) involving 4 or fewer coronary artery segments (limited disease). Conversely, risk is similarly high in patients with nonobstructive disease if more than 4 segments are affected (extensive disease) compared with patients with obstructive disease (≥50% stenosis).
_
The atherosclerotic disease burden is a powerful predictor of outcomes for patients without history of coronary artery disease, facilitated by the ease of coronary calcium scanning, which approximates the total coronary atherosclerotic disease volume. However, it is infrequently performed in patients with established coronary artery disease; thus, in this population, plaque burden data are more limited. Risk characteristics and the need for assessment may conceivably differ in patients with established coronary artery disease compared with those with history of acute coronary syndromes. Aside from calcium scanning, plaque burden assessment is technically difficult, and most available data were derived from IVUS imaging. Atherosclerotic plaque burden assessment using computed tomographic angiography has recently become feasible, but long-term outcome data are not yet available. Aggregate data from IVUS-derived plaque burden assessment reveal strong predictive power for outcomes in patients with established coronary artery disease. Conventional angiographic data for atherosclerotic disease burden are similarly predictive of patient outcomes and superior to myocardial ischemia testing in an analysis of the COURAGE (Clinical Outcomes Utilizing Revascularization and Aggressive Drug Evaluation) study. Thus, the risk associated with plaque burden applies to both asymptomatic patients without prior cardiac events and those with established, stable coronary artery disease.
_
Conclusions of this review:
Despite major advancements in coronary artery imaging and identification of atherosclerotic lesion morphology associated with rupture, there is no conclusive evidence that individual plaque assessment better predicts acute coronary event risk than established risk factors, such as the extent and severity of coronary artery disease. Pathology and clinical studies consistently demonstrate that atherosclerotic plaques rupture without clinical symptoms much more frequently than is widely acknowledged, challenging the notion of a close association between plaque rupture and clinical events. Conversely, the atherosclerotic disease burden is a consistent, strong predictor of adverse cardiovascular events and deserves greater attention. Current data suggest that rather than focusing on individual coronary arterial lesions, we need a comprehensive, integrative approach for identifying and managing patients at risk of adverse cardiovascular events.
______
______
Figure below shows why Vulnerable Plaques outnumber Cardiac Events:
- Vulnerable plaques heal and stabilise
- Even if they do rupture most plaque rupture events are sub-clinical
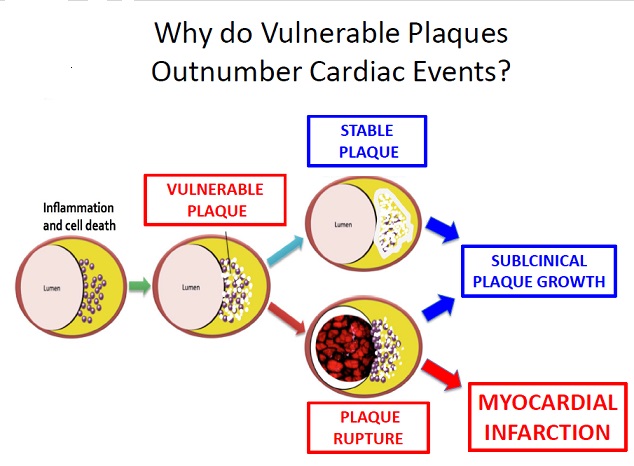
______
______
Requiem for the ‘vulnerable plaque, a 2015 study:
The notion of the ‘vulnerable plaque’ arose from autopsy studies that disclosed some two-thirds to three-fourths of fatal acute myocardial infarctions resulted from a fracture of the plaque’s fibrous cap that engendered thrombosis. The elegant post-mortem studies of pathologist pioneers redirected the cardiology community from confusion about the causality of thrombosis in ACS and a focus on vasospasm towards plaque rupture. However compelling, the number of ruptured plaques resulting in luminal occlusion in these autopsy studies lacked a ‘denominator’. While such studies could interrogate the culprit of a fatal myocardial infarction, they did not determine how many plaques with morphologic characteristics associated with vulnerability did not cause a fatal rupture.
Challenges to the ‘vulnerable plaque’ concept:
- Thin-capped, lipid-rich atheromata are not solitary, rather often multiple, and affect several arterial beds in the same individual.
- Thin-capped, lipid-rich atheromata most often persist for years without causing a clinical event.
- The risk profile and demographics of acute coronary syndrome patients are shifting worldwide (global burden, younger patients, more women, more insulin resistance/diabetes, more hypertriglyceridemia, and less low-density lipoprotein excess).
- Statin treatment and other preventive measures have begun to modify atherosclerotic disease.
- ST segment elevation myocardial infarction wanes as non-ST segment elevation myocardial infarction waxes.
- Plaque rupture declines as a cause of acute coronary syndromes while superficial erosion appears on the rise.
- Plaques underlying cerebrovascular events reveal more ‘stable’ fibrous characteristics compared with that 10 years ago.
_
Superficial erosion versus fibrous cap rupture:
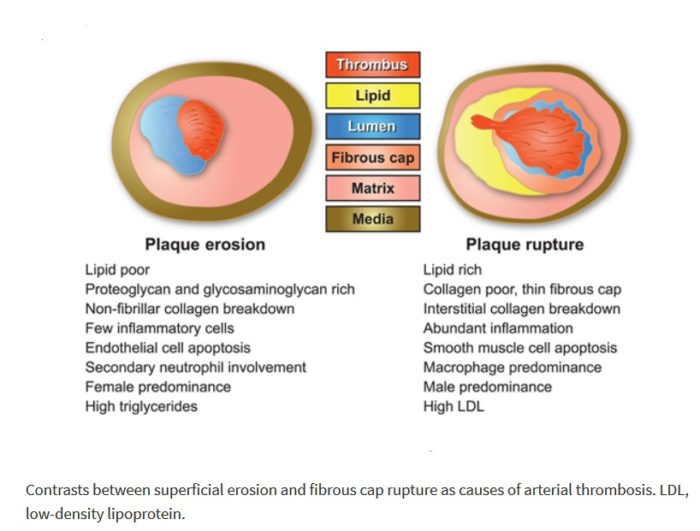
_
More recent lines of evidence suggest that plaques with thin fibrous caps and large lipid pools actually seldom rupture and cause clinical events. Multiple ‘active’ plaques often reside in the coronary and other arteries. Intravascular imaging studies in humans using ultrasound or optical coherence tomography have proved particularly illuminating. Thin-capped plaques do not inevitably rupture and cause thrombotic events. Contemporary data do not support the ‘vulnerability’ of TCFA, and indeed plaques of other morphologies may also give rise to thrombotic events as discussed below. ‘Virtual histology’, a technique based on the analysis of information gleaned from intravascular ultrasound, while incompletely validated, has provided an important challenge to the ‘vulnerable plaque’ concept. In the Providing Regional Observations to Study Predictors of Events in the Coronary Tree (PROSPECT) study, only ∼5% of thin-capped plaques as defined by virtual histology caused coronary events during a 3.4-year follow-up period. As longitudinal intravascular imaging studies such as PROSPECT enrolled higher risk patients, thin-capped plaques in lower risk populations may cause even fewer thrombotic events. Thus, the vast majority of so-called ‘vulnerable plaques’ does not exhibit clinical ‘instability’ and indeed seldom provokes ACS. Moreover, the consequences of a plaque disruption depend not only on the ‘solid state’ of the atheroma itself, but also on the fluid phase of blood, for example the concentrations of fibrinogen, endogenous inhibitors of fibrinolysis, and pro-coagulant microparticles.
_
We currently find ourselves amidst a shift in disease manifestations of atherosclerosis due to altered demographics, attendant changes in risk factor profiles, efforts to control tobacco abuse, and the increased prevalence of statin treatment. Myocardial infarction has ‘gone global’, not only affecting predominantly middle-aged Caucasian males in higher socioeconomic strata, and no longer associates mainly with cigarette smoking, hypertension, and high low-density lipoprotein (LDL). Women, non-Caucasians, younger individuals, and those with obesity, insulin resistance or frank diabetes, and high triglycerides and low high-density lipoprotein (HDL) represent an increasing proportion of our patients with ACS. We also now find ourselves the midst of a transition in the presentation of ACS, with STEMI on the wane and non-ST segment elevation myocardial infarction (NSTEMI) rising. While much of the increase in NSTEMI might result from the introduction of ever more sensitive troponin assays, shifting ACS previously classified as ‘unstable angina’ to NSTEMI, the decline in STEMI, and rise in NSTEMI began before the use of such assays. The temporal decline in STEMI incidence accompanies a substantial decline in stroke incidence and case fatality. These findings strengthen our proposition regarding a transition in the pathological mechanisms and presentations of the acute complications of atherosclerotic disease.
_
Concomitant with this trend from STEMI dominance towards NSTEMI, statin use has risen. While the temporal coincidence of a shift in ACS pathogenesis and the penetration of statin therapy do not prove causality, substantial evidence supports such a relationship. Animal studies show that lipid-lowering and/or statin treatment can reinforce the fibrous cap, decrease the lipid pool, and reduce inflammation. Human imaging studies buttress the notion that statin therapy reduces the lipid content of plaques and augments the proportion of the plaque composed of fibrous tissue, a characteristic associated with resistance to rupture. Studies on retrieved atherosclerotic plaque specimens in the Athero-Express collection have shown a time-dependent shift in the morphology of human atherosclerotic plaques over the last dozen years or so. Plaques obtained from more recent patients with symptomatic carotid artery disease reveal significantly more fibrous, non-inflammatory characteristics. This temporal trend towards plaques with morphologic characteristics of ‘stability’ in biobanked plaques also applied to asymptomatic patients. Surprisingly, statin use only explains part of this shift towards more fibrous lesions. Thus, other determinants merit careful consideration, including public policies that reduce passive smoking. Analysis of the histologic features of >1500 plaques showed that large atheroma with high macrophage content have significantly declined from 2002 to 2011, supporting our contention that the classical notion of the ‘vulnerable plaque’ has receded in relevance. Although obtaining plaque suitable for analysis from human coronary arteries presents challenges, the change in characteristics observed in human carotid atheroma likely sheds light on the current STEMI/NSTEMI transition.
_
Is the dominant mechanism of coronary thrombosis shifting from plaque rupture to superficial erosion?
The oft-quoted autopsy studies that classified culprit lesions of ACS morphologically found that rupture of the fibrous cap more commonly accounted fatal acute myocardial infarction than superficial erosion. Work from several leading pathologists has identified superficial erosion as a cause of ACS and highlighted the characteristics of lesions associated with fatal erosion that actually contrast quite starkly with those attributed to ruptured plaques (see figure above). As opposed to lesions associated with plaque rupture, those that underlie superficial erosion do not have thin fibrous caps. They harbor fewer inflammatory cells. They lack large lipid pools. Rather than insufficient interstitial collagen, lesions that cause superficial erosion accumulate abundant extracellular matrix, notably proteoglycan and glycosaminoglycans. Superficial erosion occurs more commonly in women, in individuals with diabetes, and the elderly. This clinical profile reflects in many ways the changing demographics of individuals who present with ACS today. As noted above, lipid lowering, in particular statin treatment, and perhaps less active or passive smoking, may modify the characteristics of the atherosclerotic plaque in a way that thickens the fibrous cap, reduces lipid accumulation, dispels inflammation, and shrinks the volume of the lipid core. These morphologic changes should lead to ‘stabilization’ of plaques, reducing their risk of rupture.
Might a shift towards superficial erosion vs. rupture as a mechanism of thrombosis contribute to the decline in STEMI and concurrent rise in NSTEMI, or to decreased death following some strokes? While this conjecture remains speculative, emerging evidence provides hints that favour this hypothesis. Contemporary optical coherence tomography studies have shown not only a growing proportion of ACS due to erosion vs. fibrous cap rupture, but also provide preliminary evidence that erosion associates more frequently with NSTEMI than STEMI (Figure below).
_
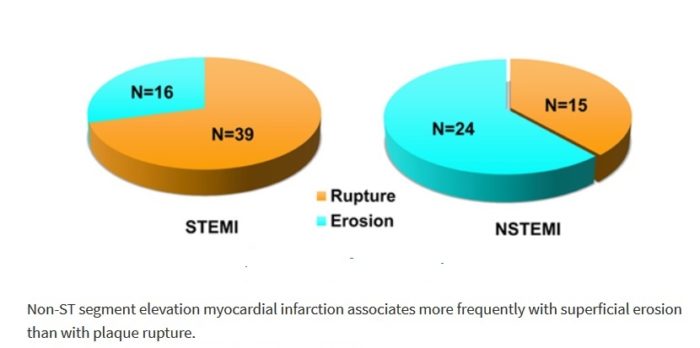
_
Clinical implications and the importance of the evolving mechanisms of acute coronary syndromes:
While plaque rupture has dominated our thinking about type 1 myocardial infarction for decades, current data should spur us to consider the shifting demographics, epidemiology, and pathophysiology of the ACS to confront this growing worldwide scourge most effectively. Despite the measures that modify so-called ‘vulnerable plaques’, a considerable burden of residual risk remains even in the statin era. The statins may have contributed to this evolution, and the introduction of new therapies that reduce LDL even further may provide yet more benefit. The shift in the risk factor profile in our ACS patients today highlights the need for novel therapies. Those that address HDL function, triglyceride-rich lipoproteins, and inflammation exemplify targets beyond LDL that may address the risk factor profile of those susceptible to residual events in the statin era.
_____
_____
Figure below summarizes arguments for and against vulnerable plaque detection attempts:
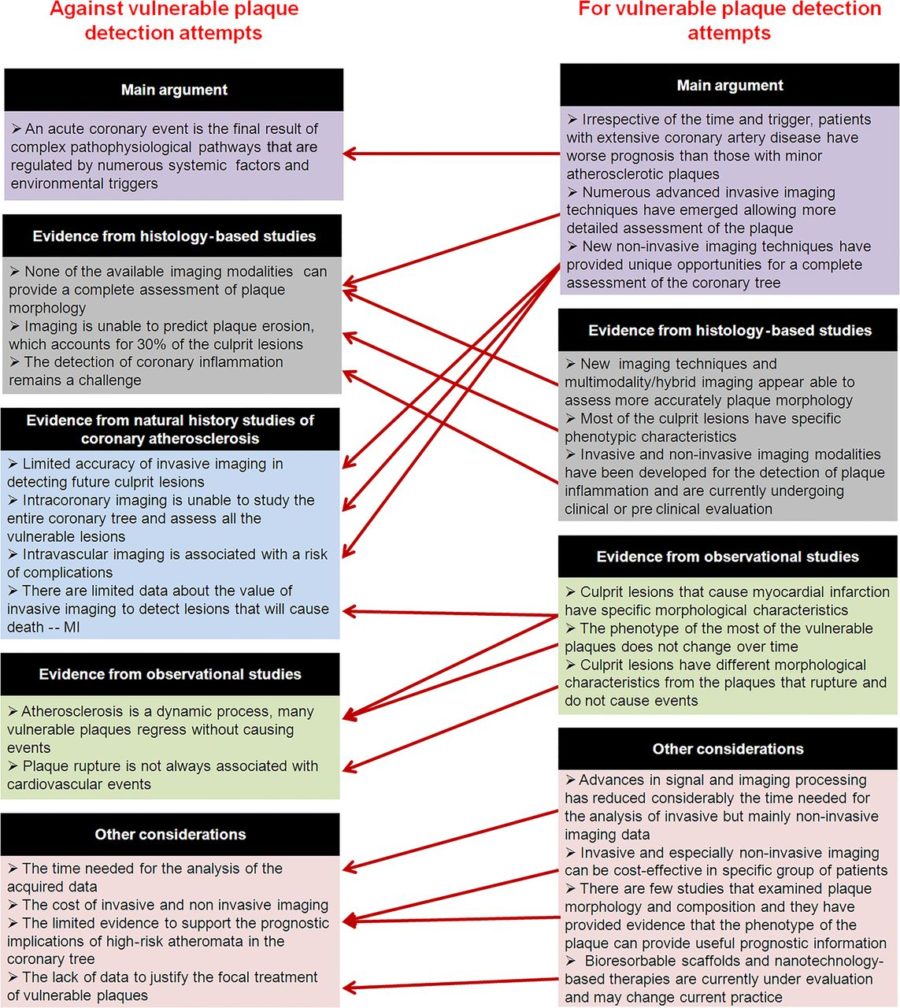
Critics of vulnerable plaque concept:
Peter Libby, professor of medicine at Harvard Medical School, has been a vocal critic of the ongoing usefulness of the vulnerable plaque concept. In a 2015 journal article, Requiem For The Vulnerable Plaque (vide supra), Professor Libby said that expending further effort into identifying a single “vulnerable’ lesion” would be a fool’s errand in light of current knowledge. Professor Libby claims that plaque rupture is much less common than previously thought. “Thin-capped plaques do not inevitably rupture and cause thrombotic events. Contemporary data do not support the ‘vulnerability’ of [thin-cap fibroatheroma], and indeed plaques of other morphologies may also give rise to thrombotic events,” he wrote. In fact, one study found only around 5% of thin-capped plaques caused a coronary event over a 3.4 year follow up, he said.
Professor Stephen Nicholls, deputy director of the South Australian Health and Medical Research Institute, said the focus was shifting from the vulnerable plaque to the vulnerable patient. Speaking at the recent Cardiac Society of Australia and New Zealand conference, Professor Nicholls said that when you look at the so-called culprit lesions in patients with an acute coronary syndrome, you’re lucky if half of them actually showed evidence of plaque rupture. “Even when the thin-cap fibroatheroma might indicate that you were at a high risk of having the event, the event was just as likely to occur in another artery. This illustrates that the vulnerable lesion doesn’t necessarily have to be where all the action is,” he told The Medical Republic. “Instead, it reflects the patient who ultimately is more vulnerable and will be more likely to benefit from more aggressive intervention,” he said.
In an article called The Vulnerable Plaque “Hypothesis”: Promise, But Little Progress, renowned cardiologist Dr Steven Nissen criticised the underlying assumptions of the hypothesis. There is no evidence that any specific plaque characteristic is associated with a poorer prognosis, and even if we assumed that a specific thin-cap fibroatheroma was the vulnerable lesion, it has limited clinical use, he argues. A 2007 study found PCI didn’t provide any benefit in terms of risk of death, myocardial infarction or other major cardiovascular events when compared to medical therapy in patients with stable coronary artery disease.
Patients are not likely to have just one plaque either. Research suggests that, when they are present, these lesions are present throughout the coronary arteries. Not only did most of these plaques never rupture, but clinicians might be surprised to find they also often disappeared, Professor Nicholls said. “A report done from virtual histology in Japan found that for every 25 so-called thin-cap fibroatheromas, when you come back and look at the same artery in the next 12 months, most of them have disappeared. In fact, the overwhelming majority of them have disappeared – if you’re lucky, 20% are still there.”
Professor MacIsaac added that, in any case, just looking at coronary artery lesions in asymptomatic patients was not very predictive of who was going to have a heart attack and when. “We have the problem that we know what groups of people are vulnerable, but when it comes to specific patients, we don’t know what’s going to happen tomorrow or the next day. We could see someone at high risk or low risk but we don’t have a way of saying ‘you’re going to have a heart attack next week and we’ve got to do something to prevent it’,” he said.
“It is clear that putting a stent in one part of one coronary artery doesn’t prevent someone necessarily from having a future heart attack,” he said.
__
Defender of the Faith:
James Muller, MD, is one of the founders of the vulnerable plaque hypothesis. He started a company, Infraredx, that uses both near-infrared spectroscopy and intravascular ultrasound to identify vulnerable plaque, which he said is the “most advanced device so far.” Muller disagreed with Nissen that ruptured lipid cores are ubiquitous. The gold standard of histology demonstrates that these lesions are not everywhere. Two large trials now underway may help settle the matter, said Muller. The 1,500 patient Lipid-Rich Plaque Study (LRP) will test whether the Infraredx device can predict future events and the 900 patient PROSPECT 2 will test the use of a bioresorbable stent in patients with a vulnerable plaque. But it should be noted that the first study is observational and the second study is not powered to evaluate clinical outcomes.
_______
_______
Genetic Susceptibility to Atherosclerosis:
Atherosclerosis, the primary cause of coronary artery disease (CAD) and stroke, is a disorder with multiple genetic and environmental contributions. Genetic-epidemiologic studies have identified a surprisingly long list of genetic and nongenetic risk factors for CAD. However, such studies indicate that family history is the most significant independent risk factor. Many Mendelian disorders associated with atherosclerosis, such as familial hypercholesterolemia (FH), have been characterized, but they explain only a small percentage of disease susceptibility (although a substantial fraction of early CAD). Most cases of myocardial infarction (MI) and stroke result from the interactions of multiple genetic and environmental factors, none of which can cause disease by itself. The contribution of genetics to the variability of atherosclerosis risk is estimated as 50%. Recent genome-wide association studies have led to the identification of over 50 gene variants which modulate atherogenesis. Risk factors for atherosclerosis are also partly genetically determined and some of the variants which play a role in atherogenesis overlap with those modulating its risk factors.
_
Although environmental factors such as diet or smoking play an important role in atherosclerosis development, genetic factors represent consequential determinant of atherosclerotic cardiovascular disease risk. Advances in techniques of molecular genetics have revealed that genetic disorders significantly influence susceptibility to atherosclerotic vascular diseases. A large number of candidate genes, genetic polymorphisms, and susceptibility loci associated with atherosclerotic diseases have been identified in recent years and, their number is rapidly increasing. Genes contribute to both the cause and the pathogenesis of virtually all abnormalities of human physiology and behaviour, including, of course, atherosclerosis. A variety of genetic factors, in addition to well studied errors of lipid metabolism, clearly predispose to atherosclerosis. Few genes apart other than those involved in lipid metabolism have such an overwhelming impact that they may be identified on the basis of family history. However, genes that predispose to hypertension and diabetes mellitus; control arterial diameter, reactivity and branching angles; affect platelet adhesiveness, thrombosis and fibrinolysis; and regulate endothelial and smooth muscle function can all be considered candidate genes for study in families predisposed to atherosclerosis. Atherosclerosis, the primary cause of coronary heart disease (CHD), involves numerous cell types and organs and a number of disparate physiological processes. It is not surprising, then, that the genetic basis of CHD is complex. This complexity is clearly illustrated by studies of rodent models; for example, experiments with transgenic and gene-targeted mice have revealed >100 genes that can influence the development of atherosclerotic lesions.
______
______
Vulnerable Plaque Formation from Obstruction of Vasa Vasorum by Homocysteinylated and Oxidized Lipoprotein Aggregates Complexed with Microbial Remnants and LDL Autoantibodies, a 2009 study:
Little attention has been paid to the function of lipoproteins as part of a nonspecific immune defense system that binds and inactivates microbes and their toxins effectively by complex formation. Because of high extra-capillary tissue pressure, aggregates of such complexes may be trapped in vasa vasorum of the major arteries. This complex formation and aggregation may be enhanced by hyperhomocysteinemia, because homocysteine thiolactone reacts with the free amino groups of apo-B to form homocysteinylated low-density lipoprotein (LDL), which is subject to spontaneous precipitation in vitro. Obstruction of the circulation in vasa vasorum, caused by the aggregated complexes, may result in local ischemia in the arterial wall, intramural cell death, bursting of the capillary, and escape of microorganisms into the intima, all of which lead to inflammation and creation of vulnerable plaques. The presence of homocysteinylated LDL and oxidized LDL stimulates production of LDL autoantibodies, which may start a vicious circle by increasing the complex formation and aggregation of lipoproteins. The content of necrotic debris and leukocytes and the higher temperature than its surroundings give the vulnerable plaque some characteristics of a micro-abscess that by rupturing may initiate an occluding thrombosis. This suggested chain of events explains why many of the clinical symptoms and laboratory findings in acute myocardial infarction are similar to those seen in infectious diseases. It explains the presence of microorganisms in atherosclerotic plaques and why bacteremia and sepsis are often seen in myocardial infarction complicated with cardiogenic shock. It explains the many associations between infections and cardiovascular disease. And it explains why cholesterol accumulates in the arterial wall. Some risk factors may not cause vascular disease directly, but they may impair the immune system, promote microbial growth, or cause hyperhomocysteinemia, leading to vulnerable plaques.
According to author’s hypothesis, infection is the cause of atherosclerosis.
Figure below shows Flow Chart for development of atherosclerotic plaque as a result of infection:
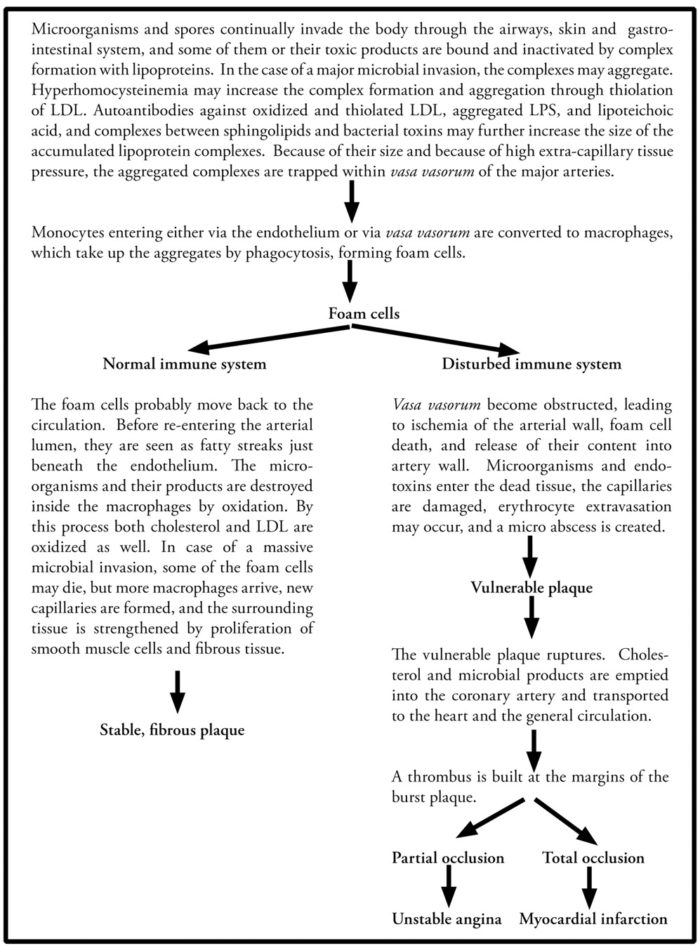
_
According to authors, LDL-cholesterol does not enter the artery through the endothelium as widely accepted, but via the capillary web of vasa vasorum in and around the arterial walls. Oxidation of LDL does not take place before LDL has entered the macrophage but occurs after phagocytosis, as part of a normal physiological process explaining why attempts to prevent cardiovascular disease by antioxidants have been largely unsuccessful. Some reasons for considering the vulnerable plaque to be a type of micro-abscess are that more than one plaque may occur simultaneously, and their temperature is higher than that of the surrounding tissue. Whereas neutrophilic polymorphonuclear leukocytes, the hallmark of pyogenic infections, are rare in stable plaques, they are always found in and around the core of vulnerable plaques, and there are just as many neutrophils in the intact as in the ruptured plaques, contradicting the assumption that their presence is secondary to rupture. Authors interpretation try to explain the clinical and laboratory similarities between myocardial infarction and myocarditis, and it explains the frequent occurrence of bacteremia and sepsis in myocardial infarction complicated with cardiogenic shock. It explains why fever, diaphoresis, leukocytosis and elevation of inflammatory markers in the blood, including CRP, the classical symptoms of an infectious disease, are common findings in myocardial infarction. Chronic elevation of CRP in patients with atherosclerosis is a risk factor for myocardial infarction. Their interpretation agrees with the almost constant finding of polymorpho-nuclear leukocytes in the myocardium in acute myocardial infarction, as well as in infarctions of other organs. It also explains a recent report of Chlamydia pneumoniae antigens within cardiomyocytes of patients with fatal myocardial infarction, an observation needing corroboration from future studies. Fatty streaks are not necessarily the precursors of atherosclerotic plaques. Fatty streaks are present in the fetus and are more frequent in early than late childhood, presumably reflecting a normal and reversible response to infections.
______
______
My Theory of atherosclerotic cardiovascular disease:
Atherosclerotic plaque rupture/erosion occurs as a result of the interactions between external mechanical triggers and vulnerable regions of the plaque when forces acting on the plaque exceed its tensile strength. Factors leading to stress on the plaque cap have been identified (i.e. blood pressure, excursion between systolic and diastolic pressure, pulse, endothelial shear stress, arterial compliance etc.) and control of these factors could be effective treatments to prevent plaque rupture/erosion. Plaque tissue properties determine the mechanical strength of plaques and may be a realistic target for therapeutic intervention, thereby decreasing the incidence of heart attack and stroke.
_______
First let me discuss cholesterol, atherosclerosis and cardiovascular disease:
_
First question:
Is atherosclerosis caused by high cholesterol?
_
‘The more LDL there is in the blood, the more rapidly atherosclerosis develops.’
This 1984 statement by the Nobel Award winners Michael Brown and Joseph L Goldstein has dominated research on atherosclerosis since then. The two men began a concerted study of the processes affecting the accumulation of cholesterol in the bloodstream. In the course of their research they discovered that low-density lipoproteins (LDL), which are primary cholesterol-carrying particles, are withdrawn from the bloodstream into the body’s cells by receptors on the cells’ surface. The genetic absence of these LDL receptors was found to be the cause of familial hypercholesterolemia, a disorder in which the body’s tissues are incapable of removing cholesterol from the bloodstream. The new understanding of cells receptors’ role in the regulation of cholesterol levels in the bloodstream spurred the successful use of drugs and the manipulation of diet in lowering blood cholesterol levels. According to the low‐density‐lipoprotein (LDL) receptor hypothesis, development of atherosclerosis is caused by a high concentration of LDL‐cholesterol in the blood, and lowering LDL‐cholesterol reverses, or at least retards, atherosclerosis, thus preventing cardiovascular disease. Familial hypercholesterolemia (FH) is an autosomal disorder characterized by increased levels of total cholesterol and low-density lipoprotein cholesterol. The FH clinical phenotype has been shown to be associated with increased coronary heart disease and premature death. The successful statin trials, with their substantial reduction of LDL‐cholesterol seemed to confirm the LDL receptor hypothesis.
_
Well, let us see the evidence:
_
Cholesterol does not predict degree of atherosclerosis at autopsy:
In 1936, Landé and Sperry noted that the degree of aortic atherosclerosis at autopsy of healthy individuals who had died violently, was independent on their blood cholesterol concentration analysed immediately after death. Their finding was confirmed by Mathur et al. and similar results were obtained by others. The objection that an analysis of cholesterol after death may not reflect its concentration during life was met by Mathur et al. who found that the cholesterol concentration was almost constant up to 16 h after death. Paterson et al. bypassed the problem by comparing the degree of atherosclerosis at death with the individuals’ cholesterol measured previously on several occasions. In all these studies, plots of blood cholesterol concentrations vs. the lipid content of the aorta or the coronary arteries were widely scattered.
More recent autopsy studies have found weak or inconsistent correlations between LDL‐cholesterol or total cholesterol and various measures of atherosclerosis. For instance, the most severe degree of atherosclerosis was found mainly in individuals with extremely high cholesterol, whereas small differences were seen in the rest. A correlation was found in White men but not in Black men, in men but not in women, in individuals below but not above age 80 years, and in the coronary arteries but not in the thoracic or abdominal aorta.
The weak and unpredictable correlations probably reflect bias, because most of the studies were performed on selected individuals. In such large projects, the main object of which was to study risk factors for cardiovascular disease, individuals with such diseases or with high cholesterol were preferred for post‐mortem examination, which means that the proportion of individuals with familial hypercholesterolemia must have been much larger than in the general population. As such patients have very high cholesterol and are more prone to vascular changes, their inclusion automatically creates a correlation between degree of atherosclerosis and LDL or total cholesterol. Therefore, to prove that the concentration of LDL‐cholesterol has importance in the general population, it is necessary to exclude individuals with familial hypercholesterolemia.
_
Cholesterol does not correlate with degree of coronary calcification:
Coronary artery calcification (CAC) seems to occur exclusively in atherosclerotic arteries and is absent in normal vessel wall. Degree of coronary calcification seems a good surrogate for degree of coronary atherosclerosis, because it correlates strongly with total plaque volume and obstructive coronary disease, and is a powerful predictor of clinical outcome. Nonetheless, degree of coronary calcification did not correlate with LDL level in the blood. Various reports have found a lack of association between low density lipoprotein cholesterol (LDL-C) and coronary calcification (CAC). However, several nonrandomized studies using statins have demonstrated attenuation of progression in CAC associated with LDL cholesterol reduction.
_
Second question:
Does high cholesterol cause cardiovascular disease?
_
The association between cholesterol and heart disease:
At first glance, it seems that we have substantial reasons to believe that high cholesterol and heart disease are closely linked. For one thing, we have a plausible-sounding explanation as to why they should be. This explanation posits that LDLs are the “bad” type of cholesterol, which can stick to the inner walls of your blood vessels and potentially clog them, leading to heart conditions such as atherosclerosis, angina and coronary heart disease (CHD). Whereas HDLs (high density lipoproteins), are the “good” type of cholesterol, and these have the opposite effect.
There are some reasons to suggest that this hypothesis is correct. Firstly, there is evidence of a correlation between high cholesterol/LDL levels and heart disease. The Framingham Study, a prospective cohort study beginning with over 5,000 participants, published a paper in 1977 which concluded that higher HDL levels and lower LDL levels were associated with a reduced risk of coronary heart disease. In addition to this, some medical treatments which reduce cholesterol/LDL levels also reduce your risk of heart disease. Statins, for example are effective in both lowering cholesterol levels, and reducing the rates of CHD, cardiovascular disease (CVD) and stroke.
Conflicting evidence:
This evidence seems to point towards there being a causal connection between cholesterol and heart disease. However, a closer examination of the issue may make us skeptical of this conclusion.
Firstly, the evidence about the association between high LDL levels and heart disease is more complicated than it first appears. A follow-up from the initial Framingham study suggested that there was only an increase in mortality, by heart disease or other causes, in people with higher cholesterol levels under the age of 50. This is significant if we want to offer medication or lifestyle advice about preventing heart disease to those over 50 years old. A recent systematic review even suggests that patients over the age of 60 actually lived longer if they had higher LDL levels, directly contradicting the hypothesis that you are more likely to die of heart disease the higher your cholesterol levels are.
Moreover, despite statins showing a correlation between lowered cholesterol levels and a reduced risk of heart disease, some other medications do not show this connection. Niacin, for example, is a cholesterol lowering drug which is known to decrease LDL levels (and increase HDL level), but has been shown to cause no significant reduction in risk of heart attack, stroke or mortality by heart disease. Even more worrying is the case of Torcetrapib. This experimental drug was designed to reduce rates of heart disease by lowering patient’s LDL levels (and increase HDL level), but the research was discontinued early because it was linked to an increased risk of mortality and heart disease.
_
If LDL‐cholesterol does not correlate with degree of atherosclerosis, why does a high cholesterol predict cardiovascular disease?
The answer may be that cardiovascular disease is not synonymous with atherosclerosis. A high LDL or total cholesterol may be secondary to uncontrolled factors that promote cardiovascular disease in other ways and cause hypercholesterolemia at the same time, for instance lack of physical activity, mental stress, smoking, and obesity. It is generally assumed that their effect on cardiovascular disease is mediated through the high cholesterol, but this may be a secondary phenomenon. Physical activity may benefit the cardiovascular system by improving endothelial function, or by stimulating the formation of collateral vessels; mental stress may have a harmful influence on adrenal hormone secretion, smoking increases the oxidant burden; in these all situations the high cholesterol may be an epiphenomenal indicator that something is wrong.
The role of LDL‐cholesterol for atherosclerosis growth has been exaggerated, a finding with consequences for the prevention of cardiovascular disease. For instance, as the statins exert their beneficial influence on the cardiovascular system by several mechanisms, it may be wiser to search for the lowest effective dose instead of the dose with maximal effect on LDL‐cholesterol. Neither should an elevated LDL‐cholesterol be the primary target in cardiovascular prevention.
_
Third question:
Does lowering cholesterol reduce plaque vulnerability?
_
Let me show statin studies which affect plaque volume and composition thereby stabilizing plaques and prevent ruptures:
- Uchida and co-workers performed intracoronary angioscopy in 157 patients presenting with stable angina. In a 12-month follow-up period, ACS occurred more frequently in patients with glistening yellow plaques (69%) than in those with white plaques (3%). Intracoronary angioscopy is applied as a tool for monitoring changes in plaque morphology following pharmaceutical therapy. Using this technique, Takano and colleagues were able to demonstrate an effect of preventive treatment with atorvastatin. Interestingly, lipid-lowering therapy with atorvastatin changed plaque color and morphology as determined by angioscopy, thereby suggesting plaque stabilization.
- Takarada et al demonstrated that statin therapy significantly increased the fibrous cap thickness on OCT in patients with hypercholesterolemia at 9-month follow-up. Recent OCT study showed that atorvastatin therapy at 20 mg compared with 5 mg provided a greater increase in fibrous cap thickness in coronary plaques of patients with unstable angina pectoris.
- Inoue et al. evaluated the plaque changes (plaque, low attenuation plaque and lumen volume, remodelling index) by serial coronary CT angiographies in 26 statin treated patients as compared to eight subjects who refused the statin treatment (controls). Serial CT angiographic evaluations of coronary plaques allowed for the assessment of statin treatment related reduction of the plaque and necrotic core volume.
- Ultrasmall super paramagnetic particles of iron oxide (USPIO) can be taken up by activated macrophages in vulnerable plaques and are visible on T2-weighted sequences as low signal intensity areas in MRI. The ATHEROMA trial was conducted to test the ability of USPIO to detect plaques in forty asymptomatic patients, demonstrating a significant reduction in plaque uptake with high-dose statin over a 3-month period.
- FDG-PET/CT has been used to detect the anti-inflammatory effect of short-term statin treatment on aortic lesions, in correlation with the circulating inflammatory biomarkers. In particular, a significant reduction of FDG activity was reached after atorvastatin treatment. These findings provide in vivo evidence that the medium dose of atorvastatin (40 mg) might have a beneficial effect on plaque stability in 12 weeks, suggesting that a relatively low dose and a short treatment interval are sufficient to observe changes in plaque burden. Ishii et al. prospectively assessed the effect of 6-month therapy with 5 vs. 20 mg of atorvastatin on reducing FDG uptake in aortic plaques with PET/CT, and showed that only atorvastatin 20 mg significantly reduced plaque inflammation.
- Several serial IVUS studies have shown that high-intensity statin therapy induces regression in plaque burden. This was recently confirmed by a post hoc analysis including data from 3495 patients. Kataoka et al published pooled data from 8 clinical trials on the beneficial effect of statins in IVUS-derived high-risk plaques (large atheroma volume, positive remodeling, and spotty calcification). The authors reported that atheroma regression was highest in patients with high-risk plaques, suggesting a plaque-stabilizing effect of statin therapy. Nissen et al studied the effect of intensive lipid lowering with rosuvastatin on coronary atheroma in 507 patients undergoing IVUS examination at baseline and after 2 years of follow-up. The marked reduction in LDL was associated with an 11.1% decrease in coronary plaque volume as measured by IVUS.
_
All above mentioned imaging studies consistently showed that statins reduce plaque volume and changes its composition to prevent rupture.
However, when you look at the so-called culprit lesions in patients with an acute coronary syndrome, you’re lucky if half of them actually showed evidence of plaque rupture. Pathological studies show that 30 % of ACS caused by plaque erosion. Plaques undergoing erosion do not have thin fibrous caps. They harbor fewer inflammatory cells. They lack large lipid pools. Rather than insufficient interstitial collagen, lesions that cause superficial erosion accumulate abundant extracellular matrix, notably proteoglycan and glycosaminoglycans. Superficial erosion occurs more commonly in women, in individuals with diabetes, and the elderly. These erosion associates more frequently with NSTEMI than STEMI. We also now find ourselves the midst of a transition in the presentation of ACS, with STEMI on the wane and NSTEMI rising. Concomitant with this trend from STEMI dominance towards NSTEMI, statin use has risen. While the temporal coincidence of a shift in ACS pathogenesis and the penetration of statin therapy do not prove causality, substantial evidence supports such a relationship. Most statin studies show that statins reduce plaque volume and some studies show statins change plaque composition. When plaque volume is reduced, it will increase circumferential wall stress as radius of artery is increased at plaque level. So although statins stabilize plaques, rather paradoxically, it increases mechanical stress due to blood pressure. This increased mechanical stress due to blood pressure may cause these stabilized plaques to erode. Statins may stabilize vulnerable plaque and prevent rupture but cannot prevent plaque erosion and thrombosis. In fact, statin use may make stabilized plaques undergo erosion rather than rupture. Despite the measures that modify so-called ‘vulnerable plaques’, a considerable burden of residual risk remains even in the statin era.
In studies of women and the elderly, hypercholesterolemia is a weak risk factor for cardiovascular disease, or, in most cases, not a risk factor at all, although the large majority of cardiovascular deaths occur in people above 65 years of age.
Also, when LDL baseline level is less than100 mg/dL, statins do not reduce mortality. Also, the event rate after complete revascularization [all diseased arterial systems with vessel size ≥1.5 (2.0–2.25 mm for PCI) with at least one significant stenosis >50% receive a graft (or stent)] remains relatively high with MACE occurring in 30% of patients after 5 years despite optimum statin therapy. On the top of it, various studies show that atherosclerotic plaques alternate between vulnerable and stable phenotypes spontaneously, so it is difficult to attribute plaque stability to statins per se.
_
Very Low cholesterol levels may indeed be harmful:
What you need to know first and foremost is that cholesterol is good for you. It’s present in every single cell in your body where it helps to produce cell membranes, hormones, vitamin D and bile acids to help you digest fat.
Cholesterol also helps in the formation of your memories and is vital for neurological function, that is why the finding that low cholesterol is linked to memory loss is not at all surprising. In fact, when your cholesterol levels go too low, a host of negative things happen in your body.
Researchers are still trying to find out more about the connection between low cholesterol and health risks. There is no consensus on how to define very low LDL cholesterol, but LDL would be considered very low if it is less than 40 milligrams per deciliter of blood.
Although the risks are rare, very low levels of LDL cholesterol may be associated with an increased risk of:
- Cancer
- Hemorrhagic stroke
- Depression
- Anxiety
- Preterm birth and low birth weight if your cholesterol is low while you’re pregnant
The potential risk of lowering LDL cholesterol to very low levels has not been confirmed, and its association with certain health risks is still under debate.
________
Finally, in a nutshell:
- The LDL level does not correlate with extent of atherosclerosis or CAC except at very high levels.
- The role of LDL‐cholesterol for atherosclerosis growth has been exaggerated, a finding with consequences for the prevention of cardiovascular disease. High cholesterol is correlated with mortality only under the age of 50 years in men. In studies of women and the elderly, hypercholesterolemia is a weak risk factor for cardiovascular disease, or, in most cases, not a risk factor at all, although the large majority of cardiovascular deaths occur in people above 65 years of age.
- There are many confounding variables like smoking, physical inactivity and mental stress, that are present in a person with high cholesterol which may affect atherosclerosis and cardiovascular risk independently of cholesterol levels.
- Although statins stabilize plaques and prevent ruptures, about 30 to 50 % of ACS are caused by plaque erosions where statins won’t work. In fact, statin use may make stabilized plaques undergo erosion rather than rupture.
- When LDL baseline level is less than100 mg/dL, statins do not reduce mortality.
- The event rate after complete revascularization [all diseased arterial systems with vessel size ≥1.5 (2.0–2.25 mm for PCI) with at least one significant stenosis >50% receive a graft (or stent)] remains relatively high with MACE occurring in 30% of patients after 5 years despite optimum statin therapy.
- Very Low cholesterol levels may indeed be harmful.
_______
Now let me discuss mechanical and hemodynamic stress on plaques:
[Read ‘Biomechanical factors in vulnerable plaque formation and rupture’ in earlier paragraphs]
_
Atherosclerosis is disease of the intima of the medium and large sized arteries due to deposition of LDL in intima. However medium and large sized veins are spared despite same amount of LDL in blood. The reason is blood pressure. Atherosclerosis can occur in any large or medium sized artery – but not in the veins, the vessels that carry blood back to the heart. Mean arterial blood pressure (MAP) is approximately 100mm Hg. This extreme pressure affects the inner lining of the vessels, allowing cholesterol to collect more easily. Normal pulmonary artery pressure is 8-20 mm Hg at rest, therefore pulmonary artery and its branches are rarely affected by atherosclerosis, and pulmonary artery atherosclerosis is accelerated in patients with hypertensive pulmonary vascular disease. The veins, however, operate on low pressure, so they are not as susceptible to build-up. Blood pressure in inferior vena cava is only 3-8 mm Hg. So, despite carrying the same amount of LDL in blood, veins are not susceptible to atherosclerosis due to low blood pressure. When saphenous vein grafting was done in coronary artery bypass surgery, these veins showed development of atherosclerosis as now they are subjected to high pressure. On the other hand, according to 2004 study, artery interposed to vein did not develop atherosclerosis and underwent atrophic remodeling in cholesterol-fed rabbits. Here left common carotid artery grafts, approximately 5 cm long, were placed in the right external jugular vein position of 24 New Zealand White rabbits. After surgery, rabbits were fed with high lipid diet for 1, 2, 4 and 12 weeks, respectively. Serum lipid levels were measured and the right common carotid artery and grafted left common carotid artery were harvested at above mentioned time points. Serum lipid levels were also measured in six rabbits receiving normal chow. Vessel wall thickness was measured and analyzed by image processing system. Hyperlipidemia occurred in all rabbits fed with high lipid diet. Fatty streak and atherosclerotic plaques were observed and lipid drops enriched in medial smooth muscle cells in control right common carotid arteries 4 weeks after surgery. In the grafted arteries, no fatty streak and atherosclerotic plaque were seen and the vessel wall thickness decreased continuously after surgery. Three months after surgery, grafted arteries possess similar structures as that of veins. The artery interposed to vein did not develop atherosclerosis and underwent atrophic remodeling in cholesterol-fed rabbits suggesting that local hemodynamic load (i.e. blood pressure) was the most important determinant influencing the development of atherosclerosis.
_
Circumferential wall tension due to hypertension plays a pivotal role in aorta remodelling is a 2006 study carried out to investigate the role of hypertension in the genesis and localization of intimal lesions and medial remodelling found in the prestenotic segment in relation to a severe stenosis of the abdominal aorta just below the diaphragm. Male young rats were divided randomly into operated group, animals submitted to surgical abdominal aorta stenosis, and sham-operated group, a control group of animals submitted to sham operation to simulate abdominal aorta stenosis. Aortas in the hypertensive prestenotic segment with increased circumferential wall tension were characterized by enlarged heterogeneous endothelial cells elongated in the direction of the blood flow, diffusely distributed conspicuous neointimal plaques and medial thickening. The immunohistochemical analysis revealed an increased expression of eNOS, iNOS, nitrotyrosine and transforming growth factor-β (TGF-β) in endothelial cells and/or smooth muscle cells in this segment. This study findings suggest that increased circumferential wall tension due to hypertension plays a pivotal role in the remodelling of the prestenotic segment through biomechanical effects on oxidative stress and increased TGF-β expression.
_
The impact of blood pressure variability on coronary plaque vulnerability in stable angina: an analysis using optical coherence tomography, a 2017 study:
Background:
Blood pressure variability (BPV), especially visit-to-visit BPV, has been reported to be a risk factor for cardiovascular disease. The impact of BPV on coronary plaque vulnerability remains uncertain. The aim of this study was to investigate the relationship between BPV and coronary plaque vulnerability.
Patients and methods:
From August 2013 to May 2014, 36 patients with both hypertension and stable angina pectoris who underwent a percutaneous coronary intervention guided by frequency-domain optical coherence tomography were investigated retrospectively. The size of the lipid cores and the thickness of the fibrous cap covering the lipid core were measured by frequency-domain optical coherence tomography, and authors calculated the blood pressure coefficient of variation (CV) and SD as intraindividual visit-to-visit BPV.
Results:
Both SD and CV of systolic blood pressure (SBP) correlated positively with lipid arc (SBP-SD: r=0.68, P<0.01; SBP-CV: r=0.64, P<0.01) as well as average SBP (r=0.48, P<0.01). Fibrous cap thickness did not correlate with blood pressure variables or BPV.
Conclusion:
BPV is related to coronary plaque volume, but not to coronary plaque vulnerability. In addition to conventional coronary risk factors, BPV may be a therapeutic target for coronary atherosclerosis.
_
Hypertension Enhances Advanced Atherosclerosis and Induces Cardiac Death in Watanabe Heritable Hyperlipidemic Rabbits, a 2018 study:
Hypertension is a major risk factor for the development of atherosclerosis. Cardiovascular risk has been reported to be significantly increased in hyperlipidemic patients with hypertension. However, it is not clear whether hypertension can directly destabilize plaques, thereby enhancing cardiovascular events. To examine whether hypertension enhances the development of atherosclerosis and increases plaque vulnerability, authors generated hypertensive Watanabe heritable hyperlipidemic (WHHL) rabbits by surgical removal of one kidney and partial ligation of the other renal artery and compared the nature of aortic and coronary atherosclerosis in hypertensive WHHL rabbits with normotensive WHHL rabbits. All hypertensive WHHL rabbits died from 34 to 56 weeks after surgery, whereas no normotensive WHHL rabbits died. Pathologic examinations revealed that hypertensive WHHL rabbits showed different degrees of myocardial infarction caused by severe coronary stenosis along with myocardial hypertrophy. Furthermore, aortic lesions in hypertensive WHHL rabbits exhibited a higher frequency of intraplaque hemorrhage and vulnerable plaques than those in normotensive WHHL rabbits. These results indicate that hypertension induced by the surgical removal of one kidney and partial ligation of the other renal artery method in WHHL rabbits may not only enhance the development of atherosclerosis but also destabilize the plaques, increasing cardiac death.
_
Hydrodynamic pressure is usually cited as the reason that atherosclerosis is localized only within systemic arteries. This explanation is correct because the arterial pressure damages the endothelium. LDL is non-living. It cannot infiltrate intima Suo-Moto. It is the blood pressure that pushes LDL through endothelial gaps. Higher the BP, higher the push. By the same reasoning, atherosclerotic plaques are localized to areas of the intimal surface where the hydrodynamic forces, turbulence of blood flow, and tissue pressure are especially high, namely at the branching points of arteries, within tortuous arteries, and within coronary arteries that are compressed by myocardial contractions. What actually causes these plaques to erupt or their fibrous caps to erode is still a mystery, although inflammation of the lesion is a prime suspect. When this inflammation is combined with other stresses, such as high blood pressure (increased mechanical stretching and contraction of the arteries with each heart beat), it can cause the thin covering over the plaque to split, spilling the contents of the vulnerable plaque into the blood stream. The propagating pulse wave causes cyclic changes in lumen size and shape, with deformation of plaques, particularly the ‘soft’ ones. Mechanical stretching and contraction of the artery, with each heartbeat, i.e. the pulse, also contribute to rupture of the thin covering membrane, spewing clot-promoting plaque contents into the blood stream. Vigorous exercise can induce coronary plaque rupture through several triggering mechanisms: increased wall stress due to high blood pressure or increased heart rate and plaque disruption caused by coronary artery spasms or increased flexing of diseased coronary arteries.
_
Elevated heart rate seems to play a role in the development and progression of coronary atherosclerosis, and numerous studies have shown an association between increased heart rate and cardiovascular mortality. Experimental animal studies have demonstrated a link between heart rate and the burden of both coronary and carotid atherosclerosis. An increased heart rate may play a role in the progression of atherosclerosis due to the increased frequency of the biomechanical load imposed on the vessel wall (i.e. repetitive wall strain). With every heartbeat, an atherosclerotic plaque is exposed to the arterial pressure wave, inducing wall stress, leading to repetitive plaque deformation (i.e. strain). Plaque strain may result in minor tissue damage, which can accumulate in time (crack propagation). A high biomechanical load could lead to rupture of plaque microvessels, cap fissures, and ultimately, cap rupture. Ruptured microvessels or fissures may then provide a point of entry for inflammatory cells and erythrocytes into the plaque, leading to further plaque destabilization. With a decreased heart rate, the vascular wall has a lower exposure to oscillatory tensile and wall shear stresses, decreasing the frequency of the biomechanical load. A reduction in shear and tensile stress is thought to reduce endothelial damage, which leads to a decreased endothelial permeability to inflammatory mediators. Heart rate lowering treatment leads to a reduction in vulnerable plaque features in atherosclerotic rabbits, a 2017 study, found that heart lowering treatment with Ivabradine in an atherosclerotic rabbit model is associated with a reduction in vulnerable plaque features and suggests that heart rate reduction may be beneficial for inducing or maintaining a more stable plaque phenotype.
_
HOPE and ONTARGET trials have shown a larger reduction in CVD events that could be predicted from the reduction in blood pressure, supporting plaque-stabilizing effects. Beta blockers lower circumferential wall stress via reductions in blood pressure, heart rate, left ventricular mass, and catecholamine surges that occur with stress and exertion, thereby stabilize plaques. Angiotensin converting enzyme (ACE) inhibitors lower the blood pressure and may have plaque-stabilizing effects by reducing the synthesis of angiotensin II. Endogenous angiotensin II induces atherosclerotic plaque vulnerability and elicits a Th1 response in ApoE-/- mice, a 2004 study, found that angiotensin II, within the context of hypertension and hypercholesterolemia, independently from its hemodynamic effect behaves as a local modulator promoting the induction of vulnerable plaques probably via a T helper switch.
_
Vicious cycle of blood pressure and atherosclerosis:
Blood pressure is cardiac output versus resistance especially in small vessels. Atherosclerosis predominantly affects large and medium sized arteries, which do not contribute to peripheral resistance, so it should not raise blood pressure. However, when your heart beats, it sends a surge of blood into your arteries. If your arteries are healthy and elastic, they will expand as blood travels through them. This “give” in your arteries keeps blood pressure from rising too high. If your arteries contain plaques, they will be less flexible. A strong reduction in compliance of large arteries due to severe extensive atherosclerosis is directly responsible for raising systolic blood pressure because of the impairment of the buffering function of the arteries on the cardiac pulse wave. In other words, blood pressure drives atherosclerosis and atherosclerosis raises systolic blood pressure, so vicious cycle of blood pressure and atherosclerosis continues irrespective of LDL level.
_
Invasive coronary angiography is currently the gold standard for the diagnosis of CAD and provides an accurate and detailed overview of the anatomy of the coronary artery tree, including precise quantification of the degree of stenosis. Accordingly, the technique is extensively used to guide further treatment strategies, such as coronary angioplasty or bypass surgery.
However, the evaluation of percentage diameter stenosis has limited value in predicting future cardiac events. Indeed, as demonstrated during the follow up of patients admitted for acute myocardial infarction, almost two thirds of plaques prone to rupture were located in non-flow-limiting atherosclerotic lesions, and only a minority were located in severely obstructed lesions. Although the likelihood of occlusion for an individual lesion is directly related to the severity of stenosis, non-obstructive lesions are far more common and thus may frequently cause coronary occlusion due to their greater number (Figure below). Accordingly, evaluation of the percentage diameter stenosis by means of invasive coronary angiography does not allow differentiation between stable and unstable plaques.
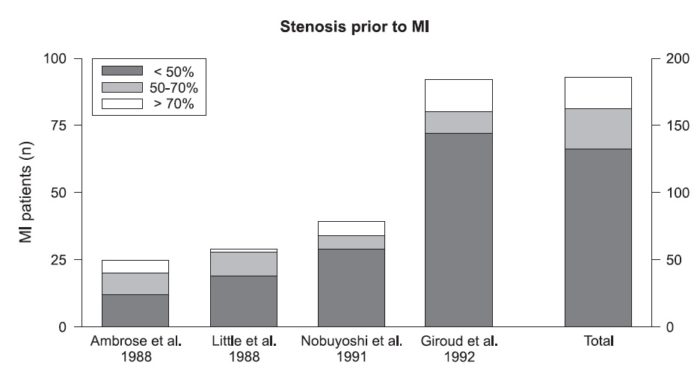
Figure above shows stenosis severity and related risk of myocardial infarction (MI) as assessed by repeated angiographic examination. As can be observed from the figure, lesions that are non-significant (<50% stenosis) on prior angiography are frequently the underlying cause of MI.
_
Counter-view study:
Another study showed that rupture incidence was highest with luminal area narrowing > 75%.
Expanded morphometric examination of ruptures and TCFAs was performed to identify relevant parameters of plaque rupture, besides cap thickness. In this series of culprit plaques, 65% of ruptures exhibited >75% cross-sectional luminal area narrowing, whereas 35% showed stenosis of <75%. The latter lesions were further subdivided into those with luminal narrowing of 50% to 75% (28% of lesions) and <50% (7% of lesion) as seen in the figure below.
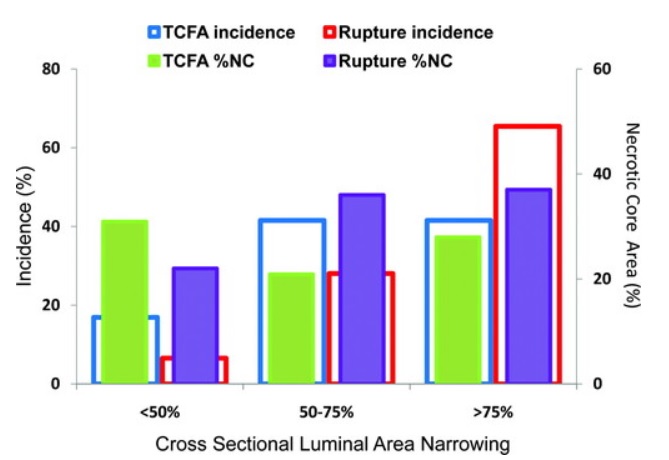
Figure above shows the relative incidence of TCFAs and ruptures (open bars) and necrotic core area (solid bars) stratified by cross-sectional area luminal narrowing into tertiles of <50% (<25% diameter stenosis), 50% to 75% (25% to 50% diameter stenosis), and >75% (>50% diameter stenosis). Note that the incidence of lesions with <75% stenosis is greater for TCFAs than for ruptures. Moreover, although the mean percentage area of necrotic core in ruptures progressively increases with stenosis, this is not the case for TCFAs.
In the clinic, a high percentage of potential vulnerable plaques with >75% stenosis, irrespective of morphology, will likely be treated using an invasive strategy, thereby removing the risk for rupture. Therefore, the majority of plaques at high risk for rupture potentially amenable to noninvasive treatment are in the target stenosis range of 50% to 70%.
So earlier studies showed almost two thirds of plaques prone to rupture were located in non-flow-limiting atherosclerotic lesions, while recent study showed that vast majority of AMI were found to have significant stenosis underneath their thrombus. In other words, studies contradict each other vis-à-vis degree of stenosis and propensity to ACS.
_
Circumferential wall tension is directly proportional to radius of vessel (and blood pressure) but shear stress is inversely proportional to cube of radius of vessel. Therefore, any lumen stenosis would reduce wall tension but increase shear stress on plaque surface. Wall tension would be more but shear stress will be less in mild stenosis as compared to severe stenosis where wall tension will be less and shear stress will be more; as wall tension and shear stress change reciprocally as lumen diameter changes.
1 pascal = 10 dyne per square centimeter
1 mmHg = 133.32239 pascal = 1333.22 dynes/cm2
100 mmHg = 133322 dynes/square centimeter
Mean arterial pressure (MAP) is approximately 100 mmHg equivalent to133322 dynes/square centimeter.
Mean circumferential wall tension (CWT) is calculated by Laplace law according to the following formula:
CWT = MAP × r, where CWT is expressed in dyne/cm, MAP is mean arterial pressure (dynes/cm2) and r is
radius of artery (cm)
Now radius of normal coronary artery is about 2 mm or 0.2 cm (diameter 4mm)
CWT = 133322 X 0.2 = 26664.4 dynes/cm
Tensile stress (TS) is computed as TS = CWT/IMT, where TS is expressed in dyne/cm2, CWT is circumferential wall tension (dyne/cm) and IMT is intima-media thickness (cm) of coronary artery wall.
Average IMT of normal coronary artery wall is about 0.4 mm (0.04 cm)
TS = CWT/IMT = 26664.4/0.04 = 666610 dynes/cm2
So circumferential wall tensile stress on endothelium is 666610 dynes/cm2 at 100mm of mean arterial pressure in a coronary artery of 4 mm diameter and 0.4 mm IMT.
Larger the lumen of artery, lesser the IMT, greater the circumferential wall stress if blood pressure is same.
Remember, wall stress = wall tension / IMT
Circumferential wall stress is proportional to circumferential wall tension but inversely proportional to thickness of artery. Therefore, the terms wall stress and wall tension cannot be used interchangeably.
_
Endothelial Shear Stress (ESS):
Endothelial cells also are subject to the frictional force shear stress generated by blood flowing past their apical surfaces. This force is determined by the mean fluid flow rate, its viscosity, and the physical dimensions of blood vessels. For Newtonian fluids (defined as those for which flow velocity does not alter viscosity) flowing in rigid containers with unvarying internal geometry, uniform velocity gradients develop such that fluid velocity is least at the stationary vessel wall and rises with increasing distance from it. Fluid flow with this characteristic uniform velocity gradient is termed laminar flow because the fluid can be viewed as a series of molecular layers (laminae) slipping past each other with increasing velocity as the center of the container is approached as seen in the figure below.
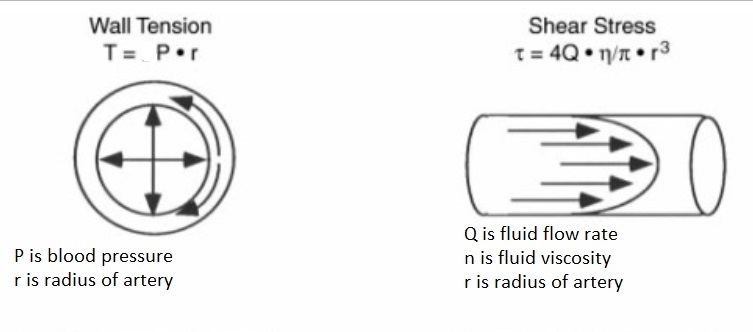
Figure above shows schematic representation of the effects of vessel wall tension and shear stress on endothelial cells. Wall tension develops in response to transmural pressure gradients and causes cell stretch, with cell deformation in all directions. Shear stress is the fluid frictional force acting at the apical surface of endothelial cells. Shear stress results in unidirectional cell deformation.
If mean flow rate is constant, the greater the resistance to flow, either because of high fluid viscosity or small vessel diameter, the greater the shear stress. Shear stress causes cell deformation (strain), which raises cytoskeletal tension, although the direction of the deformation differs from that produced by transmural pressure gradients as seen in the figure above. In the purest sense, therefore, shear stress and transmural pressure gradients are unrelated forces, each of which cause endothelial cell (EC) strain. In both cases, there is deformation of cytoskeletal elements, cell membranes, and sites of cell-cell and cell-matrix adhesion with consequent tension development. However, in the case of shear stress, the force on a given cell acts in one direction, whereas stretch due to transmural pressure gradients occurs in all directions. It is also of note that only the ECs, but not vascular smooth muscle cells or pericytes, are exposed to shear stress, whereas all vessel wall components are deformed by transmural pressure gradients.
_
The complex interactions of blood flow, viscosity and arterial geometry generate the different ESS patterns that are characterized by their direction (unidirectional, bidirectional) and magnitude (low, moderate, high). Moderate ESS varies between 1.5 -3.0 Pa over the cardiac cycle, and it is encountered in relatively straight arterial segments, where it exhibits unidirectional flow with atheroprotective effects. High ESS refers to values over 3 Pa, which is present at the stenotic site of atherosclerotic plaques. Low ESS (below 1-1.5 Pa) with undisturbed unidirectional flow occurs at the inner areas of curvatures and upstream of stenosis. Low oscillatory ESS is characterized by bidirectional flow (reverse flow), and it is observed at the ostia of branches, downstream of stenotic plaques and at the lateral walls of bifurcations. Low and/or oscillatory ESS promotes pro-atherogenic pathways according to some studies, and it is a key factor in the development and progression of focal atherosclerosis.
_
Different studies calculated different values of ESS in different degree of stenosis.
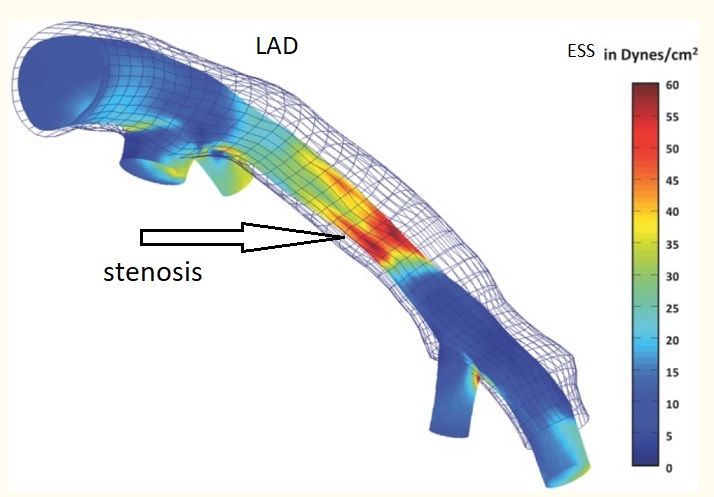
Figure above shows a study that calculated ESS > 60 dynes/cm2 at severe stenotic site in LAD artery.
_
According to Effect of Stenosis Severity on Wall Shear Stress Based Hemodynamic Descriptors using Multiphase Mixture Theory, 2018 study:
Higher the degree of stenosis, higher ESS.
| Degree of lumen stenosis in % | Average Endothelial shear stress in dynes/cm2 |
| 30 | 41 |
| 50 | 48 |
| 70 | 83 |
| 85 | 353 |
_
There is lot of confusion about stenosis measurement in coronary artery, whether lumen diameter stenosis or whether lumen area stenosis should be considered, or the worst view, i.e., the most narrowed diameter, or should we consider all the views and report the averaged value? For example, a 90% concentric lesion with a diameter of 2mm and lumen area of 3.14mm2 versus an eccentric lesion with a 90% stenosis in one view (diameter 2mm) and 0% stenosis in the second view, and diameter of 4mm, which, using a prolate ellipsoid model, produces a lumen area of 6.28mm2. Obviously, the coronary flow would be much more reduced in the former than the latter lesion. Detailed discussion is beyond the scope of this article. Most vessels are circular or elliptical, and the stenoses are frequently asymmetric when viewed in cross section. A vessel with 50% diameter stenosis in one plane is 50% stenotic. If it has 50% stenosis in two planes it is 75% narrowed. Sufficient to say that when diameter or radius is halved, it produces approximately 75% lumen stenosis.
_
Let me give one example with 75% lumen stenosis (50 % diameter/radius stenosis).
ESS will be about 60 to 80 dyne/cm2 as shown in preceding paragraph.
Now let me calculate circumferential wall tension in this coronary artery with half radius of 1 mm. Due to stenosis and plaques, IMT of this coronary artery will be 1 mm or 0.1 cm.
CWT = MAP × r, where CWT is expressed in dyne/cm, MAP is mean arterial pressure (dynes/cm2) and r is
radius of artery (cm)
Now radius of this diseased coronary artery is about 1 mm or 0.1 cm (diameter 2mm)
CWT = 133322 X 0.1 = 13332.2 dynes/cm
Tensile stress (TS) is computed as TS = CWT/IMT, where TS is expressed in dyne/cm2, CWT is circumferential wall tension (dyne/cm) and IMT is intima-media thickness (cm) of coronary artery wall.
IMT of severely stenotic coronary is about 1 mm (0.1 cm)
TS = CWT/IMT = 13332.2/ 0.1 = 133322 dynes/cm2
So circumferential wall tensile stress on endothelium is 133322 dynes/cm2 at 100mm of mean arterial pressure in a coronary artery of 2mm diameter and 1 mm IMT having 75% lumen stenosis.
As compared to normal coronary artery having wall stress of 666610 dynes/cm2 at 100 mm Hg MAP, diseased coronary artery will have 133322 dynes/cm2 wall stress with 75 % lumen stenosis at 100 mm Hg MAP.
Although circumferential wall stress on plaque has fallen due to severe stenosis, it is substantially greater than increased ESS of 80 dynes/cm2 due to severe stenosis.
In other words, the mechanical stress imposed by blood pressure is substantially greater than hemodynamic stress imposed by shear stress on endothelial surface. I mean there is no comparison. Endothelia shear stress is miniscule as compared to circumferential wall stress, and therefore all directional wall stress can easily overwhelm unidirectional shear stress. No matter whether you have plaques with mild, moderate or severe stenosis, the difference in shear stress would be too insignificant to have any impact on plaque rupture/erosion as wall stress is substantially more powerful than shear stress at any degree of stenosis. So, wall stress would determine propensity to rupture/erode and not shear stress no matter the degree of stenosis. And since wall stress is more in mild stenosis as compared to severe stenosis, plaque rupture/erosion is more likely to occur in plaques with mild stenosis as compared to severe stenosis provided plaque composition is same. The highest wall stress typically occurs at the upstream shoulders of a stenotic plaque or within a non-stenotic plaque, so these are the areas of plaque rupture/erosion.
When circumferential wall stress generated by pulse and blood pressure peak at very specific weak points depending on geometry of vulnerable plaques, fibrous cap thinness and tensile strength of collagen, rupture/erosion occurs. It is the surge in blood pressure that cause plaque rupture/erosion of vulnerable plaque, for example: exertion, vigorous exertion, anger, fear and wake-up. Of course, plaque composition and geometry (of artery and plaque) do matter but it plays second fiddle to blood pressure.
______
Finally, in a nutshell:
- Atherosclerosis is disease of the intima of the medium and large sized arteries due to deposition of LDL in intima. However medium and large sized veins are spared despite having the same amount of LDL in blood. The reason is blood pressure. LDL is non-living. It cannot infiltrate intima Suo-Moto. It is the blood pressure that pushes LDL through endothelial gaps. Higher the BP, higher the push. Blood pressure is the most important determinant influencing the development of atherosclerosis. At arterial bifurcation (e.g. carotid bifurcation), there is turbulence of blood flow in addition to blood pressure, so atherosclerotic plagues are maximal at arterial bifurcations.
- High blood pressure (increased mechanical stretching and contraction of the arteries with each heart beat) leads to higher circumferential wall stress which can cause the thin covering over the plaque to split, spilling the contents of the vulnerable plaque into the blood stream.
- The propagating pulse wave causes cyclic changes in lumen size and shape, with deformation of plaques increasing likelihood of inflamed plaques’ rupture/erosion. Elevated heart rate (pulse rate) seems to play a role in the development and progression of coronary atherosclerosis, and plaque vulnerability.
- Vigorous exercise can induce coronary plaque rupture/erosion through several triggering mechanisms: increased wall stress due to high blood pressure and increased heart rate, and plaque disruption caused by coronary artery spasms or increased flexing of diseased coronary arteries.
- Various studies have shown that larger reduction in CVD events could be predicted from the reduction in blood pressure, supporting plaque-stabilizing effects. Hypertension accelerates atherosclerosis. Changes in blood pressure can tilt a plaque from stability into instability and rupture/erosion.
- No matter whether you have plaques with mild, moderate or severe stenosis, the difference in shear stress would be too insignificant to have any impact on plaque rupture/erosion as wall stress is substantially more powerful than shear stress at any degree of stenosis. So, wall stress would determine propensity to rupture/erode and not shear stress no matter the degree of stenosis. Since circumferential wall stress is more in mild stenosis as compared to severe stenosis, plaque rupture/erosion is more likely to occur in plaques with mild stenosis as compared to severe stenosis provided plaque composition is same. When circumferential wall stress generated by pulse and blood pressure peak at very specific weak points depending on geometry of vulnerable plaques, fibrous cap thinness and tensile strength of collagen, rupture/erosion occurs. It is the surge in blood pressure that cause plaque rupture/erosion of vulnerable plaque, for example: exertion, vigorous exertion, anger, fear and wake-up. Of course, plaque composition and geometry (of artery and plaque) do matter but it plays second fiddle to blood pressure.
- Blood pressure drives atherosclerosis and atherosclerosis raises systolic blood pressure, so vicious cycle of blood pressure and atherosclerosis continues irrespective of LDL level.
__
Final conclusion:
Mechanical stress imposed on arterial intima by blood pressure and pulse rate are the main biomechanical factors causing atherosclerosis and plaque rupture/erosion, with LDL cholesterol, plaque composition and geometry (of artery and plaque) playing the second fiddle to blood pressure and pulse rate. Control of blood pressure and pulse rate would reduce cardiovascular mortality far more than LDL control by statins. I support lowering of normal blood pressure values in young and middle-aged adults to 110/70 mm Hg instead of 120/80 mm Hg.
______
______
Moral of the story:
_
- Atherosclerosis is a systemic disease of the intima of the medium-sized and large arteries due to endothelial dysfunction resulting in multifocal, smoldering, immunoinflammatory, fibro-proliferative, prothrombotic, angiogenic and multifactorial disease; characterized by formation of atherosclerotic plaques. The endothelium is responsible for vasodilation, inhibition of thrombosis, suppression of smooth muscle cell proliferation and inhibition of inflammatory responses; all these functions are affected in atherosclerosis due to endothelial dysfunction. Both the barrier function and secretory capacity of the endothelial cells are disrupted in atherosclerosis. This manifests as an increase in permeability to lipids and inflammatory cells (mainly monocytes and T lymphocytes) derived from the blood. Endothelial cells, lipids, leukocytes, and intimal smooth muscle cells are the major players in the development of this disease. During the progression of atherosclerosis, endothelial cells, macrophages, and smooth muscle cells die from apoptosis or necrosis. Atherogenesis starts early in infancy, and evolves slowly over decades, leading progressively to the formation of atherosclerotic plaques.
_
- Atherosclerotic plaques are asymmetric focal thickenings of the intima and the main components of the atherosclerotic plaque are connective tissue extracellular matrix, including collagen, proteoglycans, and fibronectin elastic fibers; cholesterol, cholesteryl esters, and phospholipids; necrotic debris, and calcium; and cells such as monocyte-derived macrophages, T lymphocytes, and smooth muscle cells. Varying proportions of these components occur in different plaques, giving rise to a spectrum of lesions. It is multifocal, in that, lesions have a tendency to occur at predictable anatomic sites of the arterial tree. It predictably occurs at bifurcations, side branches, and opposite flow dividers at areas of high or low endothelial shear stress and turbulent blood flow. There is an orderly cephalad progression over time starting with the iliacs and progressing cephalad to the aorta, coronaries, carotids and cerebral vessels. The most devastating consequences of atherosclerosis, such as heart attack (myocardial infarction MI) and stroke (cerebrovascular accident CVA), are caused by superimposed thrombosis over these plaques. If thrombosis-prone atherosclerotic plaques could be detected and thrombosis averted, atherosclerosis would be a much more benign disease.
_
- The coronary arteries are, in fact, more prone to atherosclerotic plaques than many other arteries in the human body; and atherosclerosis appears to be more prevalent in the left coronary arterial system compared to the right. There is progression from asymptomatic atherosclerosis, to a high-risk/vulnerable plaque, to a thrombosed plaque, and to clinical events. Coronary events continue to be the leading cause of death worldwide and improved preventive measures are needed because, for many individuals, sudden coronary death is the first sign of the disorder. And even those who survive an acute coronary syndrome remain at high risk despite optimum therapy.
_
- The initial trigger of atherogenesis results apparently from the hemodynamic stress, i.e. turbulent blood flow, which elicits endothelial cell activation in atherosclerosis prone areas (arterial bifurcations). In early atherosclerotic lesions, the distribution of fatty streaks in the vasculature is predominantly in areas where there are changes in blood flow such as bifurcations and back currents causing endothelial injury. Endothelial cell dysfunction is the earliest detectable physiologic manifestation of atherosclerosis. The major atherogenic risk factors, such as smoking, high low-density lipoprotein (LDL) levels, hypertension, and diabetes, have all been shown to impair endothelial function.
_
- When atherosclerosis is initiated by endothelial dysfunction, the dysfunctional endothelium exhibits an increased permeability, generates reactive oxygen species (ROS) and expresses inflammatory adhesion proteins and chemokines. The endothelium permeability allows an influx of plasma components in the subendothelial area, so low-density lipoproteins (LDL) lipoproteins infiltrate intima and subsequent oxidation of LDL molecules cause an inflammatory response with infiltration of T-lymphocytes, and macrophages that consume LDL and form foam cells. Smooth muscle cells (SMCs) migrate from the media to the intima where they proliferate, uptake modified lipoproteins and secrete extracellular matrix (ECM) proteins that stabilizes the plaques (fibrous cap). This is initially protective, but with further LDL accumulation, macrophage cell death is ultimately triggered, contributing to further inflammation and the development of a necrotic core of soft unstable atheroma. Plaque inflammation also triggers smooth muscle cell loss and the production of matrix metalloproteinases (MMP) that weaken the fibrous cap, predisposing it to plaque rupture. The thick necrotic acellular lipid core becomes increasingly hypoxic, stimulating angiogenesis, with the formation of immature microvessels prone to intra-plaque hemorrhage (IPH). Numerous local and systemic factors determine whether the lesion continues to progress or heal. If the lesion progresses, it develops a necrotic center, with innumerable dead and/or dying macrophages, which are associated with a rim of microcalcifications. When the cap separating the atheroma from the vessel lumen is ≤65 microns thick, the atheroma is likely to rupture, hence, the term “vulnerable plaque”. If the plaque ruptures, the interaction of the necrotic core material and blood cause a thrombus. If the thrombus is small and plaque less occlusive, it is likely that the event remains subclinical, although it might contribute to the plaque growth. In contrast, if the thrombus is nearly or totally occlusive, especially with a luminally stenotic plaque, a clinical event becomes likely.
_
- Culprit plaque is a plaque responsible for coronary occlusion resulting in clinical event (myocardial infarction and death). Vulnerable plaque is a future culprit plaque. The term vulnerable plaque refers to all asymptomatic atherosclerotic plaques which are at risk of thrombosis or rapid progression to become culprit lesions, thereby places the patient at risk for future major adverse cardiac events (MACE). A vulnerable plaque is not necessarily a soft plaque, a noncalcified plaque, an AHA type IV plaque, or a non-stenotic plaque. The concept of vulnerable plaque has been proposed as a paradigm to improve the prevention of cardiovascular disease by identifying atherosclerotic plaques at higher risk for causing future cardiovascular events. Vulnerable plaques can develop not only in native coronary arteries but also in the neointima after long-term coronary stent implantation.
_
- The morphological traits typically associated with rupture-prone plaques are found in lesions usually referred to as thin-cap fibroatheroma (TCFA). They include a large eccentric lipid-rich necrotic core occupying approximately one-quarter of the plaque area, a thin fibrous cap less than 65 μm heavily infiltrated with macrophages and other inflammatory cells, spotty calcifications, positive remodeling and vasa vasorum proliferation. So TCFA is a rupture-prone vulnerable plaque. Non-rupture prone vulnerable plaques include plaques that undergo superficial erosion or plaques that protrude calcified nodules in the lumen. These non-rupture prone vulnerable plaques are also at a risk of thrombosis similar to rupture-prone TCFA.
_
- Inflammation appears to be a pan-coronary process, resulting in “multifocal” patterns of plaque instability and vulnerability. An acute coronary syndrome may be a clinical marker of widespread (multifocal) disease activity in the coronary arteries, possibly related to inflammation. Angiographic observations demonstrate that nearly 50% of patients with acute MI manifest multiple sites of plaque instability. IVUS, angioscopic and pathological studies have confirmed that ACS patients harbor multiple sites of plaque rupture and, importantly, multiple TCFAs as well. It is important to note that inflammation exists not only locally within the coronary bed, but appears to be systemic.
_
- Vascular calcification is an established feature of atherosclerosis. The role that the calcification plays in plaque vulnerability is still under debate. Some studies indicated beneficial effects in stabilizing the plaque, making it stiffer and less prone to rupture, while others tended to believe it would increase the risk of plaque rupture. Microcalcification within the thin fibrous cap, or spotty calcification, may serve as a marker of plaque vulnerability. In contrast, larger calcified sheets consisting of merged areas of microcalcification are associated with plaque stability. Previous research has suggested that it is the soft – that is, noncalcified and lipid-laden – plaque that holds the highest risk of rupturing and triggering heart attacks. However, new research suggests that hard, calcified plaque may be a stronger indicator of adverse cardiovascular events. It is now known that the more calcium you have, the more plaques you have, and the more likely you are to have an event. Coronary artery calcium (CAC) is a disease marker and not risk marker, and it is correlated with adverse cardiovascular events no matter soft or hard plaque, no matter microcalcification or macrocalcification.
_
- The natural course of coronary plaques with features of vulnerable plaque is not unidirectional, and it does not always involve a move toward plaque rupture manifesting as ACS, but rather it often remains asymptomatic, with ruptured plaques healing. The plaque rupture might occur without being followed by the formation of a flow-limiting thrombus and the development of AMI. Coronary plaque ruptures have been shown in 7%–27% of stable angina patients and in 3% of patients dying of noncardiac causes. Not all cases of intracoronary thrombosis necessarily lead to a clinical event. Subclinical plaque rupture occurs in 11.5% of patients with stable CAD and 21.5% in patients who presented with ACS. There is wide variation in composition from plaque to plaque, even within a single patient, i.e. patients may have plaques that are fibro-calcific as well as lipid-rich. They may have plaques that are TCFAs, and they may have plaques that are inflamed and ruptured. There are also transitions in the life of a plaque, in which it becomes eroded or frankly disrupted over time; intra-plaque hemorrhage is also an important pathophysiological process that contributes to plaque progression, instability and possibly calcification. Longitudinal imaging studies have suggested that plaque morphology may change over time, and many suspected vulnerable plaques may not progress over the course of years and may even regress to more stable lesions. Actually, the vast majority of thin-capped, lipid-rich atheroma persists for years without causing any clinical event. To simplify, the TCFA can be viewed as an earlier stage of the plaque, whereas the fibro-calcific lesion is a more chronic scarred-down spectrum of the process. Most ruptures and clotting events are too small to produce symptoms. The clot organizes and contracts over time, leaving behind narrowing(s) called stenoses. These narrowing(s) are responsible for the symptoms of the disease and are identified, after the fact, by the changes seen on stress tests and angiography, and treated with bypass surgery or angioplasty with or without stents. In lesions with advanced lumen narrowing (>50% stenosis), histopathology almost invariably reveals one or more healed subclinical plaque ruptures.
_
- Coronary arteries may respond to plaque growth by expansion or shrinkage of the vessel wall, defined as positive and negative remodeling, respectively. Positive remodeling is an outward compensatory remodeling in which the arterial wall grows outward in an attempt to maintain a constant lumen diameter, while negative remodeling is defined as a local shrinkage of vessel size. Whereas vulnerable coronary plaques are generally associated with positive remodeling, stable plaques are characterized by negative remodeling of the vessel wall. Because artery walls typically enlarge in response to enlarging vulnerable plaques, these plaques do not usually produce much stenosis of the artery lumen. Therefore, they are not detected by cardiac stress tests or angiography, the tests most commonly performed clinically with the goal of predicting susceptibility to future heart attack. Positive remodeling in vulnerable plaque rupture mechanics explain why many heart attacks occur without prior symptoms.
_
- Diabetes mellitus increases the prevalence of plaque formation and rupture as diabetes mellitus affects various cellular and molecular effectors involved in plaque development and rupture. Insulin is a known activator of endothelial nitric oxide synthase (NOS), leading to increased production of NO, which has potent antiatherogenic effects. Insulin has protective effects against atherosclerosis at physiological and pharmacological concentrations, although better glycemic control and other metabolic parameters make it difficult to attribute anti-atherogenic effects of insulin treatment to insulin per se.
_
- Stable plaques plus larger coronary arteries that dilate more in exercise should be highly protective against coronary events in high volume exercisers. Regular exercise decreases the risk of coronary heart disease but in men with severe coronary artery disease, sudden death related to strenuous exercise is associated with acute plaque rupture. Exercise is good for you, but is there a point at which strenuous exercise is either no longer beneficial or perhaps even harmful? The issue is not resolved yet.
_
- The coronary plaque phenotype, which is independently associated with non-culprit lesion–related MACE, cannot be predicted by clinical and angiographic findings, emphasizing the potential value of more comprehensive plaque characterization (with either intravascular or non-invasive imaging) for more accurate risk stratification.
_
- For primary prevention, emphasis is on non-invasive imaging of vessels in those deemed to be at higher risk after initial assessment with demographic characteristics and plasma markers. Non-invasive techniques might allow us to rapidly screen low-moderate risk patients and therefore to prevent acute coronary syndrome (ACS). For secondary prevention in patients already undergoing catheterization, invasive imaging can be performed with minimal added risk and cost, and has considerable advantages over non-invasive imaging as high microscopic resolution is possible to study the plaque composition. Among all imaging techniques, intravascular ultrasound (IVUS) and optical coherence tomography (OCT) provide the closest information to match the histopathology of atherosclerotic plaques. However, the additional time and cost, risk of complications, increased time required to process the obtained data, and lack of evidence about how we should treat high-risk patients make unrealistic the regular application of 3-vessel invasive imaging in clinical settings. Non-invasive imaging such as CT carries a great potential because it overcomes the abovementioned limitations of intravascular imaging modalities.
_
- From a clinical standpoint, multi-detector coronary CT angiography has emerged as the best non-invasive imaging modality in terms of assessing both lumen area and plaque composition, throughout most of the coronary vasculature (vessels >1.5 mm in diameter, where the majority of plaques are located). Its accuracy in detecting lesion presence is noninferior to IVUS, and consequently, it exhibits an excellent rule‐out ability with a negative prognostic value approaching 100%, even in prolonged follow‐up. Furthermore, although unable to quantify cap thickness, it is nevertheless able to detect presence of thin cap fibroatheromas, comparing favorably with OCT. Several CT defined plaque characteristic have now been described to reliably identify unstable plaques. Large plaque volume, low CT attenuation, napkin-ring sign, positive remodeling, and spotty calcification are all associated with a high risk of acute cardiovascular events. Retrospective studies have demonstrated the utility of these plaque features to predict future acute coronary events. If these can be confirmed in prospective studies, the intrinsic benefits of non-invasive imaging will position coronary CT angiography firmly in our armamentarium to image coronary arteries and help prevent acute coronary events.
_
- Atherosclerotic plaques of coronary arteries can be classified into two types according to color by coronary angioscopy. Yellow plaques are frequently observed at the site of a culprit lesion in the setting of acute coronary syndromes. These plaques have thin fibrous caps with lipid-rich cores and inadequate collagen content. In contrast, white plaques have thick fibrous caps or are completely fibrous. Various studies suggest that yellow plaques are more vulnerable to rupture than white plaques.
_
- The landmark PROSPECT study has confirmed two key principles that will be relevant as we continue to innovate and improve our treatment of cardiovascular disease.
First, angiographic guidance alone is not a good predictor of future events; grayscale intravascular ultrasound is much better than angiography at determining the severity of plaque, and which blockages are most likely to cause future events. It showed that plaque burden ≥ 70%, minimal luminal area ≤ 4 mm2 and TCFA characteristics on IVUS were independently associated with subsequent major adverse cardiovascular events (MACE) derived from non-culprit lesions; and all of these characteristics were invisible to the coronary angiography but easily identifiable by IVUS.
Second, all coronary artery disease is not the same, and differences in underlying tissue type can in fact change the risk profile of a lesion. Some blockages are more active and can quickly progress to a clinical event. Other blockages are more stable, and when combined with proper medical therapy, have an extremely low risk of causing a future event.
_
- Experts agree that no single modality has emerged as optimal for imaging vulnerable plaque. In fact, it may be a hybrid approach that ultimately meets the goal of imaging vulnerable plaque. Hybrid PET/CT enables measurement of functional and structural features of atherosclerosis, in which FDG uptake detects plaques with an ongoing inflammatory process, whereas CT identifies chronic calcification as a manifestation of the late stage of the diseases.
_
- All eligible vulnerable plaque imaging techniques, whether non-invasive or invasive, have demonstrated relatively superior negative predictive value, but the positive predictive values remain too low to warrant a pre-emptive intervention. Also, the imaging techniques need to study the serial behavior of the vulnerable lesions over time as vulnerable lesion may remain as vulnerable lesion or stabilizes with time.
_
- Many inflammatory biomarkers and enzymes have been suggested as potential biomarkers of vulnerable plaques but there are no useful, scientifically proven biomarkers of vulnerable plaque currently available.
_
- Although plaque ruptures and erosions are indeed responsible for most culprit lesions in patients with acute events, because the frequency of subclinical plaque ruptures is vastly underestimated, the assumption that identifying lesions prone to rupture will prevent acute coronary events is unrealistic. Of the many plaque ruptures occurring in patients with atherosclerotic disease, very few will trigger symptomatic events, rendering it exceedingly difficult to predict adverse outcomes associated with particular lesions. The quest for the vulnerable plaque rests on the erroneous assumption that detecting coronary atherosclerotic lesions, which are prone to rupture or erode, will identify individuals at high risk of suffering acute coronary events. Not all vulnerable plaque ruptures and not all plaque ruptures lead to a heart attack or stroke. Studies have demonstrated that during a heart attack, several arteries are often involved with plaque rupture, yet not all develop occlusive thrombosis. There is strong, consistent evidence that plaque ruptures occur frequently without causing events. Indeed, ruptured plaques typically heal clinically silently and lead to plaque progression & growth, increased plaque burden and subsequent lumen narrowing. It is only the exceptional case of plaque rupture or erosion that leads to an event, a perfect storm scenario. Given the very low absolute risk associated with individual plaques, the critical question is what intervention would be beneficial in this setting—even if identification of such plaques would be possible at low risk and costs.
_
- Despite vulnerable plaque features presence having high sensitivity in cases of acute events, prospective studies, such as Providing Regional Observations to Study Predictors of Events in the Coronary Tree (PROSPECT), VH‐IVUS in Vulnerable Atherosclerosis (VIVA), and the European Collaborative Project on Inflammation and Vascular Wall Remodeling in Atherosclerosis (ATHEROREMO‐IVUS), have consistently shown that their positive predictive value is low, implying low prognostic value on an individual basis. When studies suggest that plaques may alternate between vulnerable and nonvulnerable phenotypes, it casts doubt on the significance of diagnosing plaque vulnerability and, conversely, suggest that nonvulnerable plaques may transition into an unstable morphology and suffer rupture or erosion. So even stable, by all accounts, plaques can rupture/erode.
_
- The atherosclerotic disease burden, its metabolic activity, and response of coagulation system to plaque disruption are important predictors of acute coronary event risk and deserve our attention more than individual plaques. The focus on imaging and treatment of specific vulnerable plaques fails to address the fact that atherosclerosis is a systemic, multifactorial disease, and optimal care likely consists of systemic and multifocal diagnosis and therapy, of which local plaque therapy may be a component. There is overwhelming evidence to show that atherosclerotic disease burden is a powerful predictor of outcome and not individual VP. The risk of MI or death associated with individual TCFAs or vulnerable plaques is only 0.06% per year, much smaller than what is conventionally considered high risk. The risk is most strongly conveyed by the extent of coronary artery disease, but not necessarily by individual lesions, even when highly obstructive. Patients with high-grade coronary artery stenoses may conceivably carry an increased risk of myocardial infarction and death because these lesions are markers for advanced atherosclerotic disease in the coronary tree. The nonobstructive coronary artery disease may be associated with similar risks of myocardial infarction and death if it affects a larger number of coronary arterial segments. It is shown that in many cases the clot responsible for an acute coronary event occurs in a plaque that is < 50% stenotic. Therefore, the assumption that only highly stenotic sites seen on angiography are at risk for thrombotic occlusion and subsequent myocardial infarction, whereas those coronary arteries that do not contain obstructive stenosis (< 50%) are nearly free of risk for thrombotic occlusion is not valid. It is the extent of coronary artery disease mirrored by total plaques burden that determine the clinical event and not degree of luminal stenosis. The risk associated with plaque burden applies to both asymptomatic patients without prior cardiac events and those with established, stable coronary artery disease.
_
- Imaging ‘vulnerable’ plaques is a fascinating research tool providing useful insights into the pathophysiology of ACS. At present, it has little clinical use. Local therapy (angioplasty/stent), in the absence of documented ischaemia, is unproven because no randomized trial has shown that benefits of stenting vulnerable plaques exceed the complications, which include periprocedural infarction, restenosis, and stent thrombosis. Although local treatment of an individual vulnerable plaque is appealing, it fails to address the fact that atherosclerosis is a systemic disease.
_
- Coronary heart disease is a systemic illness which requires a comprehensive approach. We know that control of blood pressure, lipids, blood glucose, smoking cessation, exercise and diet have a profound effect on patient outcome. Assessing the total burden of atherosclerotic disease and its metabolic activity as well as considering systemic risk, such as inflammation, is more likely to aid in the management of patients with coronary heart disease than fixating on individual plaques.
_
- The atherosclerotic plaque composition is consistent with the intake of certain dietary atoms. Diet containing fish products is associated with stability of atherosclerotic plaque as compared to diet containing potatoes, wheat, oats and rice.
_
- Cholesterol crystals (CC) initiate inflammation in plaque and eventually destabilize the plaque. Aspiration of culprit coronary artery obstruction during PCI for AMI showed presence of large CC clusters, and it was associated with increased inflammation (IL-1β), increased arterial narrowing, and diminished reflow following PCI. An optical coherence tomography study found that presence of CC in coronary plaque was significantly correlated with a greater rate of 1-year MACE. Thus agents that dissolve or prevent cholesterol crystals formation may stabilize vulnerable plaques. When LDL infiltrates intima, it is taken up by macrophages which form foam cells which upon rupture release cholesterol crystals. HDL reverse transport cholesterol out of intima. In other words, any agent that reduce LDL level and/or increase HDL level would stabilize plaque.
_
- Both primary and secondary prevention trials have unequivocally proved that reduction in LDL cholesterol using statins reduces subsequent clinical events on average by 30%–40%. Statins significantly increased the TCFA fibrous cap thickness and reduced the lipid core arc. The mortality benefit of LDL-lowering therapy by use of statins is well established. Several serial IVUS/OCT studies have shown that high-intensity statin therapy induces regression in plaque burden. Many meta-analyses showed that high intensity statin therapy can significantly decrease the size of atherosclerotic plaques (plaque volume). Those with a high-risk plaque experienced much more benefit in terms of atheroma regression from statins than those without. This confirmed the expectation that high-risk plaques had the machinery to progress the disease, but also that the biology could be modified by interventions. It is still not clear how low LDL should be reduced, as it is only one of the risk factors. There are patients who develop MI and stroke who have perfectly normal LDL levels. Thus, there may be a threshold below which little additional benefit is seen. Whether that level is 100 or 70 mg/dL is still subject to question. When baseline LDL level is less than 100 mg/dL, statins are not reducing mortality, challenging pleiotropic actions of statins.
_
- Although plaque rupture remains the most prominent cause of ACS, it should be noted that therapeutics aimed at preventing plaque rupture (stabilizing plaques) have no effect on the incidence of ACS as a result of plaque erosion or calcified nodule. Contemporary optical coherence tomography studies have shown that not only a growing proportion of ACS are due to plaque erosion (not rupture), but also provide preliminary evidence that erosions associate more frequently with NSTEMI than STEMI. As opposed to lesions associated with plaque rupture, those that underlie superficial erosion do not have thin fibrous caps. They harbor fewer inflammatory cells. They lack large lipid pools. Rather than insufficient interstitial collagen, lesions that cause superficial erosion accumulate abundant extracellular matrix, notably proteoglycan and glycosaminoglycans. Superficial erosion occurs more commonly in women, in individuals with diabetes, and the elderly. Statins may stabilize vulnerable plaque and prevent rupture but cannot prevent plaque erosion and thrombosis. In other words, statins cannot prevent about 30 to 50 % ACS. Prevention of plaque erosion need novel therapies like targeting HDL function and triglyceride-rich lipoproteins.
_
- Our current understanding is driven by the recognition that potentially life-threatening events are caused not only by vascular changes but also by alterations in the nature of the circulating blood and the myocardium. As such, these three components—vulnerable vessel, vulnerable blood, and vulnerable myocardium—characterize what is today referred to as the vulnerable patient, who is defined as a patient with a high risk of experiencing a cardiac event. Such a patient is likely to have vulnerable blood (prone to thrombosis), vulnerable myocardium (prone to arrhythmia), and ≥1 vulnerable plaque. The consequences of a plaque disruption depend not only on the ‘solid state’ of the atheroma itself, but also on the fluid phase of blood, for example the concentrations of fibrinogen, endogenous inhibitors of fibrinolysis, and pro-coagulant micro particles. The strongest predictors of adverse events are the magnitude and activity of the coronary atherosclerotic plaque burden and the number of risk factors for a prothrombotic milieu. Prothrombotic risk factors include CRP, dyslipidemia, platelet count, homocysteine, physical inactivity, tobacco, diabetes and genetic predisposition. The recognition that cardiovascular risk is determined not only by the vulnerable vessel but also by components of the vulnerable blood and the vulnerable myocardium has major implications for future research and therapeutic developments.
_
- Although established risk factor-based screening fails miserably to predict major adverse cardiovascular events (MACE), there is no conclusive evidence that individual plaque assessment predicts MACE better than established risk factors. In fact, established risk factors correlate well with atherosclerotic disease burden and it is the atherosclerotic disease burden that is a strong predictor of adverse cardiovascular events in association with prothrombotic milieu; and both involve genetic factors as well as environmental factors. Although environmental factors such as diet or smoking play an important role in atherosclerosis development, genetic factors represent consequential determinant of atherosclerotic cardiovascular disease risk. The contribution of genetics to the variability of atherosclerosis risk is estimated as 50%. In the same way, physical inactivity and smoking may be environmental factor for prothrombotic milieu, and genes regulating platelet adhesiveness, thrombosis and fibrinolysis would contribute to genetic factors for prothrombotic milieu. The sole reason why traditional risk factor-based screening fails miserably to predict major adverse cardiovascular events is unaccounted genetic factors contributing to atherosclerosis and prothrombotic milieu.
_
- According to my theory of atherosclerotic cardiovascular disease, mechanical stress imposed on arterial intima by blood pressure and pulse rate are the main biomechanical factors causing atherosclerosis and plaque rupture/erosion, with LDL cholesterol, plaque composition and geometry (of artery and plaque) playing the second fiddle to blood pressure and pulse rate. Control of blood pressure and pulse rate would reduce cardiovascular mortality far more than LDL control by statins. I support lowering of normal blood pressure values in young and middle-aged adults to 110/70 mm Hg instead of 120/80 mm Hg.
Reasons in support of my theory:
-1. Cholesterol level does not predict degree of atherosclerosis at autopsy.
-2. Cholesterol level does not correlate with degree of coronary calcification.
-3. The role of LDL‐cholesterol for atherosclerosis growth has been exaggerated.
-4. High cholesterol is correlated with mortality only under the age of 50 years in men. In studies of women and the elderly, hypercholesterolemia is a weak risk factor for cardiovascular disease, or, in most cases, not a risk factor at all, although the large majority of cardiovascular deaths occur in people above 65 years of age.
-5. There are many confounding variables like smoking, physical inactivity and mental stress, that are present in a person with high cholesterol which may affect atherosclerosis and cardiovascular risk independently of cholesterol levels.
-6. Although statins stabilize plaques and prevent ruptures, about 30 to 50 % of ACS are caused by plaque erosions where statins won’t work. In fact, statin use may make stabilized plaques undergo erosion rather than rupture.
-7. When LDL baseline level is less than100 mg/dL, statins do not reduce mortality.
-8. The event rate after complete revascularization [all diseased arterial systems with vessel size ≥1.5 (2.0–2.25 mm for PCI) with at least one significant stenosis >50% receive a graft (or stent)] remains relatively high with MACE occurring in 30% of patients after 5 years despite optimum statin therapy.
-9. Very Low cholesterol levels may indeed be harmful.
-10. Atherosclerosis is disease of the intima of the medium and large sized arteries due to deposition of LDL in intima. However medium and large sized veins are spared despite having the same amount of LDL in blood. The reason is blood pressure. LDL is non-living. It cannot infiltrate intima Suo-Moto. It is the blood pressure that pushes LDL through endothelial gaps. Higher the BP, higher the push. Blood pressure is the most important determinant influencing the development of atherosclerosis. At arterial bifurcation (e.g. carotid bifurcation), there is turbulence of blood flow in addition to blood pressure, so atherosclerotic plagues are maximal at arterial bifurcations.
-11. High blood pressure (increased mechanical stretching and contraction of the arteries with each heart beat) leads to higher circumferential wall stress which can cause the thin covering over the plaque to split, spilling the contents of the vulnerable plaque into the blood stream.
-12. The propagating pulse wave causes cyclic changes in lumen size and shape, with deformation of plaques increasing likelihood of inflamed plaques’ rupture/erosion. Elevated heart rate (pulse rate) seems to play a role in the development and progression of coronary atherosclerosis, and plaque vulnerability.
-13. Vigorous exercise can induce coronary plaque rupture/erosion through several triggering mechanisms: increased wall stress due to high blood pressure and increased heart rate, and plaque disruption caused by coronary artery spasms or increased flexing of diseased coronary arteries.
-14. Various studies have shown that larger reduction in cardiovascular events could be predicted from the reduction in blood pressure, supporting plaque-stabilizing effects. Hypertension accelerates atherosclerosis. Changes in blood pressure can tilt a plaque from stability into instability and rupture/erosion.
-15. No matter whether you have plaques with mild, moderate or severe stenosis, the difference in endothelial shear stress would be too insignificant to have any impact on plaque rupture/erosion as circumferential wall stress is substantially more powerful than shear stress at any degree of stenosis. So, circumferential wall stress would determine propensity to rupture/erode and not shear stress no matter the degree of stenosis. Since circumferential wall stress is more in mild stenosis as compared to severe stenosis, plaque rupture/erosion is more likely to occur in plaques with mild stenosis as compared to severe stenosis provided plaque composition is same. When circumferential wall stress generated by pulse and blood pressure peak at very specific weak points depending on geometry of vulnerable plaques, fibrous cap thinness and tensile strength of collagen, rupture/erosion occurs. It is the surge in blood pressure that cause plaque rupture/erosion of vulnerable plaque, for example: exertion, vigorous exertion, anger, fear and wake-up. Of course, plaque composition and geometry (of artery and plaque) do matter but it plays second fiddle to blood pressure.
-16. Blood pressure drives atherosclerosis and atherosclerosis raises systolic blood pressure, so vicious cycle of blood pressure and atherosclerosis continues irrespective of LDL level.
______
Dr. Rajiv Desai. MD.
March 26, 2019
_____
Postscript:
I apologize for criticizing statins in my article ‘Imitation Science’. Statin therapy to reduce cardiovascular mortality is real science albeit imperfect. Blood pressure is the main power that drives atherosclerosis and plaque rupture/erosion, lipids play second fiddle to blood pressure.
_____
Footnote:
I have seen so many patients on statins with poorly controlled hypertension, and it appears that both doctors and patients are obsessed with cholesterol, and probably inadvertently ignore optimum blood pressure control. This article may help them overcome their cholesterol obsession.
_____
154 comments on “VULNERABLE PLAQUE”
Leave a Reply

You actually make it seem so easy with your presentation but I find this topic to be actually something which I think I would never understand. It seems too complex and extremely broad for me. I am looking forward for your next post, I will try to get the hang of it!|
I have read so many articles on the topic of the blogger lovers but this paragraph is in fact a pleasant paragraph, keep it up.|
Im thankful for the post.Much thanks again. Much obliged.
Looking forward to reading more. Great post.Much thanks again. Fantastic.